

9120 氨基酸分析指导原则
氨基酸分析系指采用适宜的方法测定蛋白质、多肽或其他药物制剂中氨基酸组成和/或含量。蛋白质和多肽的氨基酸分析,需要将样品先水解成游离的氨基酸才能进行分析,游离氨基酸通常需要衍生化后才能测定。
游离氨基酸的测定方法主要有柱前衍生反相色谱法、柱后衍生离子交换色谱法、离子色谱-积分脉冲安培检测法、液相色谱-蒸发光散射检测法、柱前衍生气相色谱法、衍生化毛细管电泳法、液相色谱-质谱联用法、超临界流体色谱法等,其中柱前衍生反相色谱法和柱后衍生离子交换色谱法为药品中氨基酸测定常用方法。
本指导原则概述了药品中氨基酸分析的基本要求,介绍了蛋白质和多肽样品的水解方法、常用的衍生化氨基酸测定方法及有关数据处理等内容,为药品中氨基酸的分析提供指导。
1 基本要求
1.1 仪器
氨基酸分析使用的仪器通常是高效液相色谱仪或氨基酸分析仪。柱前衍生的氨基酸通常使用高效液相色谱法分离检测;对于柱后衍生的氨基酸,由于离子交换分离过程的复杂性和对柱后衍生化反应装置的特殊要求等,一般使用商品化的氨基酸分析仪。
1.2 内标物
为了减少实验误差,氨基酸分析常采用内标法,所使用的内标物应是非天然存在的一级氨基酸,易于获取且价格便宜,在水解过程中保持稳定,其衍生物的色谱响应与浓度呈线性关系,且与待测氨基酸能有效分离。
常用的内标物包括正亮氨酸、α-氨基丁酸、正缬氨酸、肌氨酸和硝基酪氨酸等。内标物应在水解或衍生化反应前添加到氨基酸混合物中,以消除由于水解、衍生化、取样、进样、溶液稳定性和色谱条件变化等导致的差异。
1.3 方法验证
本指导原则中列出的氨基酸测定方法是指导性的方法,用户可以根据自己的实验室条件及分析品种的特点,参考本指导原则所列举的方法建立针对具体品种的适宜的氨基酸测定方法,也可以建立本指导原则未列举的其他氨基酸测定方法。对建立的氨基酸测定方法,应根据分析方法验证指导原则(指导原则9101)进行方法学验证,证明方法对所分析样品的适用性。
2 蛋白质和多肽样品的水解
蛋白质或多肽样品中的氨基酸是以结合形式存在,必须经过水解处理,形成游离氨基酸后才能进行氨基酸测定。
蛋白质或多肽的水解方法主要采用酸水解,同时辅以碱水解。其中,酸水解中使用最广泛的是盐酸水解,其产生的氨基酸不消旋,但该方法会使一些氨基酸被全部或部分破坏,如色氨酸被全部破坏,丝氨酸、苏氨酸和半胱氨酸被部分破坏,门冬酰胺和谷氨酰胺脱酰胺分别转化为门冬氨酸和谷氨酸。对于这些氨基酸可采用较特殊的处理方法或者使其转变为稳定的形式,然后再进行盐酸水解。
蛋白质或多肽样品的水解需在水解管或一次性使用的水解管中进行,常用的蛋白质和多肽的水解方法如下。
方法1 盐酸水解法
盐酸水解法又可分为液态和气态两种方式。液态水解时样品溶于水解溶液。气态水解时样品不接触水解溶液,而以水解加热过程中产生的气态酸来水解干粉或干燥后的样品,这可将来自水解溶液的污染降低至最小程度,适用于仅有少量样品的微量分析。该水解方法不适用于蛋白质或多肽样品中色氨酸、含硫氨基酸(半胱氨酸、胱氨酸和甲硫氨酸)的定量。
在一定浓度的盐酸溶液中加入适量苯酚可用于防止酪氨酸的卤化。
水解溶液:6mol/L盐酸溶液(含0.1%~1.0%苯酚)。
液态酸解: 取干燥后的蛋白质或多肽样品约3mg,置水解管中,加水解溶液1ml,用氮气置换水解管中的空气,封管。110℃水解24小时或150℃水解1小时,蒸干或减压干燥水解的样品,供分析测定用。
气态酸解: 取干燥后的蛋白质或多肽样品约1mg,置水解管中,将含有样品的水解管放入装有约1ml水解溶液的容器中,用氮气置换容器中的空气,密封容器。110℃水解24小时。取出水解管减压干燥水解后的样品,供分析测定用。
如果蛋白质在上述条件下水解不完全,可延长水解时间至48小时或72小时。
方法2 氢氧化钠水解法
本方法仅适于蛋白质或多肽样品中色氨酸的测定。
水解溶液:5mol/L氢氧化钠溶液。
水解方法: 取干燥的蛋白质或多肽样品约10mg,置聚四氟乙烯水解管中,准确加入5mol/L氢氧化钠溶液4.0ml,用氮气置换水解管中的空气后,封管,110℃水解20小时,加入6mol/L盐酸3.5ml中和至中性,用水或适宜的稀释剂适当稀释,供分析测定用。
方法3 过氧甲酸氧化酸水解法
在蛋白质或多肽水解之前,用过氧甲酸氧化样品中的半胱氨酸或胱氨酸和甲硫氨酸,使其转化为稳定的磺基丙氨酸和甲硫氨酸砜,防止半胱氨酸或胱氨酸和甲硫氨酸在水解过程中被破坏,得到的转化产物按方法1再进行盐酸水解。该水解方法不适用于蛋白质或多肽样品中色氨酸和酪氨酸的含量测定。
氧化溶液:无水甲酸-30%过氧化氢(9∶1)溶液(临用前制备并在制备后放置1小时后使用)。
水解方法: 取蛋白质或多肽约500μg,置水解管中,减压干燥,加无水甲酸20μl, 50℃加热5分钟,放冷,加氧化溶液100μl,混匀,放置10~30分钟。减压干燥去除样品中过量的试液,照方法1进行盐酸水解。
方法4 二硫代二乙酸或二硫代二丙酸还原酸水解法
半胱氨酸或胱氨酸与二硫代二乙酸或二硫代二丙酸反应后,转化为稳定的S-硫代乙酸-半胱氨酸或S-硫代丙酸-半胱氨酸,可防止半胱氨酸或胱氨酸在水解过程中被破坏,得到的转化产物按方法1再进行盐酸水解。该水解方法不适用于蛋白质或多肽样品中色氨酸的含量测定。
还原溶液: 取二硫代二乙酸(或二硫代二丙酸)适量,溶于0.2mol/L氢氧化钠溶液中,使成每1ml含二硫代二乙酸(或二硫代二丙酸)约10mg的溶液。
水解方法: 取蛋白质或多肽约20μg,置水解管中,减压干燥,加入还原溶液5μl,加异丙醇10μl,减压干燥除去所有液体,照方法1再进行盐酸水解。
方法5 双(1,1-三氟乙酰氧基)碘苯还原酸水解法
在蛋白质或多肽水解之前,用双(1,1-三氟乙酰氧基)碘苯(BTI)还原样品中的门冬酰胺和谷氨酰胺残基,使其分别转化为二氨基丙酸和二氨基丁酸残基,得到的转化产物按方法1再进行盐酸水解。该水解法适用于在门冬氨酸和谷氨酸存在下,蛋白质或多肽样品中门冬酰胺和谷氨酰胺的定量。
在离子交换色谱分离中,α,β-二氨基丙酸和α,γ-二氨基丁酸与赖氨酸间不能完全分离,因此本法测得的门冬酰胺和谷氨酰胺含量为未经BTI还原的样品与经BTI还原的样品水解后得到的门冬氨酸与谷氨酸含量之差。由于BTI还原反应影响苏氨酸、甲硫氨酸、半胱氨酸、酪氨酸的含量测定结果,如果要测定蛋白质或多肽中这些氨基酸组成,则必须通过水解未经BTI还原反应的样品来测定。
还原溶液:10mmol/L三氟乙酸溶液(溶液A)、5mol/L盐酸胍-10mmol/L三氟乙酸溶液(溶液B)、含36mg/ml BTI的二甲基甲酰胺溶液(溶液C,临用前制备)。
水解方法: 取蛋白质或多肽约200μg,置水解管中,减压干燥,加还原溶液A或溶液B 2ml和溶液C 2ml,在减压状态下密封水解管,60℃避光加热4小时,溶液用水透析去除过量的试剂,再用相同体积的正丁酸乙酯提取3次,冻干,得到的冻干粉末按方法1再进行盐酸水解。
3 氨基酸测定法
除苯丙氨酸、酪氨酸和色氨酸等少数氨基酸外,大部分氨基酸无发色基团或发光基团,在紫外检测器(UV)或荧光检测器(FLD)无响应,需要在特定条件下与衍生剂经化学反应,生成发色基团或发光基团,用紫外检测器(UV)或荧光检测器(FLD)检测。衍生化反应可分为柱前衍生和柱后衍生:前者是先将氨基酸混合物衍生化,再用反相色谱法分离后检测;后者是先用离子交换色谱法将氨基酸混合物分离,再经在线衍生化后检测。
柱前衍生化通常每次分析需要的氨基酸总量为0.5~1.0μg。
柱后衍生化通常每次分析需要的氨基酸总量为5~10μg。
氨基酸测定方法的系统适用性要求,在各品种项下规定。常用的衍生化氨基酸测定方法如下。
方法一 柱前PITC衍生氨基酸测定法
原理 氨基酸与异硫氰酸苯酯(PITC)反应,生成有紫外吸收的苯氨基硫甲酰氨基酸衍生物(PTC-氨基酸),PTC-氨基酸经反相液相色谱分离后用紫外检测器在254nm波长处检测,在一定的浓度范围内(1~1250pmol)其响应值与氨基酸浓度成正比。
特点 该法具有仪器配置要求不高、实验成本低、分离效果好等优点,适用于含氨基酸种类较多的样品分析。胱氨酸或半胱氨酸衍生物不稳定,无法定量;色氨酸与鸟氨酸的分离困难,不适合同时含有色氨酸和鸟氨酸样品的含量测定;除PTC-胱氨酸不稳定外,其他PTC-氨基酸衍生物溶液在室温条件下至少可稳定24小时。
色谱条件 用十八烷基硅烷键合硅胶为填充剂(4.6mm×250mm, 5μm);以乙腈-0.1mol/L醋酸钠溶液(取醋酸钠13.6g,加水900ml溶解,用醋酸调pH值至6.5,加水至1000ml)(7∶93)为流动相A,以乙腈-水(80∶20)为流动相B,按下表进行梯度洗脱,流速为每分钟1ml;检测波长为254nm;柱温40℃。
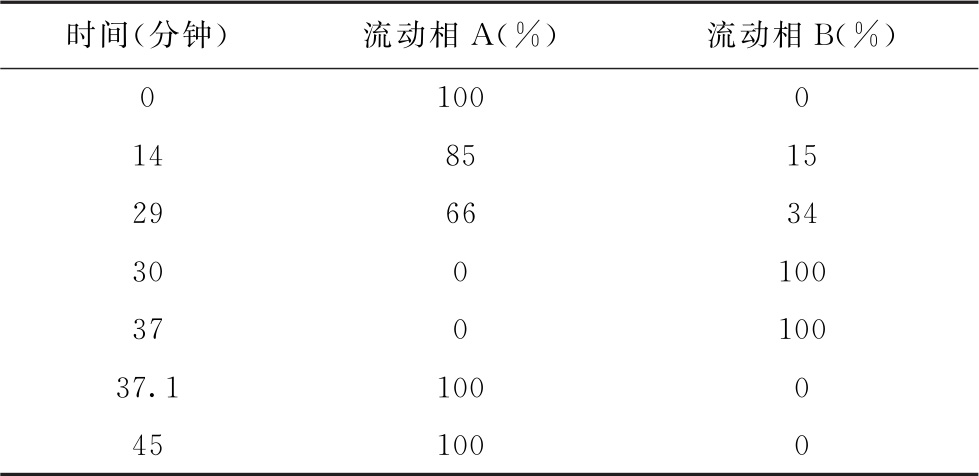
测定法 视供试品中待测氨基酸种类及其含量,取供试品、对照品及内标物(如正亮氨酸)适量,用水或0.1mol/L盐酸溶液制成含总氨基酸浓度不大于2.5mg/ml的供试品溶液和与供试品溶液浓度相当的对照品溶液。精密量取供试品溶液200μl,置离心管中,精密加入1mol/L三乙胺乙腈溶液100μl,混匀,精密加入0.1mol/L异硫氰酸苯酯乙腈溶液100μl,混匀,密封,放置1小时,加正己烷0.8ml,充分振摇20~30秒,静置分层,精密量取下层溶液2μl,注入液相色谱仪,记录色谱图;另精密量取对照品溶液200μl,自“置离心管中”起同法测定。按内标法计算供试品溶液中各氨基酸的含量。
方法二 柱前AQC衍生氨基酸测定法
原理 氨基酸与6-氨基喹啉-N-羟基琥珀酰亚胺基氨基甲酸酯(AQC)反应,生成有紫外吸收的不对称尿素衍生物(AQC-氨基酸),AQC-氨基酸经反相液相色谱分离后用紫外检测器在248nm波长处检测,在一定的浓度范围(2~100pmol)内其响应值与氨基酸浓度成正比。
特点 该法具有分离效果好、衍生物稳定等优点,适用于含氨基酸种类较多的样品分析。该法的衍生试剂价格高,AQC-氨基酸衍生物在室温下至少可稳定1周。
色谱条件 用十八烷基硅烷键合硅胶为填充剂(4.6mm×250mm, 5μm);以醋酸盐缓冲液(取醋酸铵10.8g,加水900ml溶解,用磷酸调pH值至5.0,加水至1000ml)为流动相A,以乙腈-水(60∶40)为流动相B,按下表进行梯度洗脱,流速为每分钟1.4ml;检测波长为248nm;柱温37℃。
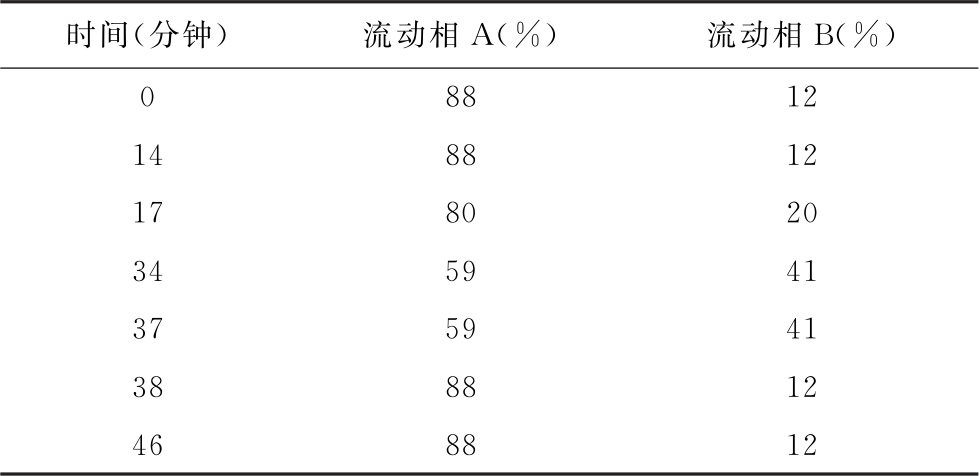
测定法 视供试品中待测氨基酸种类及其含量,取供试品、对照品及内标物(如γ-氨基丁酸)适量,用水或0.1mol/L盐酸溶液制成含总氨基酸浓度不大于0.4mg/ml的供试品溶液和与供试品溶液浓度相当的对照品溶液。精密量取供试品溶液10μl,置一小试管中,精密加入0.4mol/L pH 8.8硼酸盐缓冲液(取硼酸12.36g,加水400ml溶解,用40%氢氧化钠溶液调pH值至8.8,然后加水稀释至500ml)70μl,在涡旋状态下,精密加入AQC溶液(取AQC适量,加乙腈适量,55℃加热10分钟使溶解并稀释制成每1ml中含1.6mg的溶液)20μl,混匀,密封,55℃加热10分钟,放冷,精密量取5μl,注入液相色谱仪,记录色谱图;另精密量取对照品溶液10μl,自“置一小试管中”起同法测定。按内标法计算供试品溶液中各氨基酸的含量。
方法三 柱前OPA和FMOC衍生氨基酸测定法
原理 一级氨基酸(含伯氨基的氨基酸)在巯基试剂存在下,首先与邻苯二醛(OPA)反应,生成OPA-氨基酸;反应完毕后,加入9-芴甲基氯甲酸甲酯(FMOC),剩余的二级氨基酸(含仲氨基的氨基酸,如脯氨酸)与FMOC继续反应,生成FMOC-氨基酸;两次反应生成的氨基酸衍生物经反相高效液相色谱分离后用紫外光检测器在338nm和262nm波长处分别检测,在一定的浓度范围(25~2500pmol)内,氨基酸衍生物的吸光度与氨基酸浓度成正比。
特点 该法具有可自动化柱前衍生、实验成本低、分离效果好等优点,适用于含氨基酸种类较多的样品分析。二级氨基酸的衍生重复性较差及色谱柱使用寿命较短;OPA-氨基酸衍生物不稳定,衍生反应结束后需立即进样分析;衍生化操作也可由具有自动衍生功能的进样器完成。
色谱条件 用十八烷基硅烷键合硅胶为填充剂(4.6mm×200mm, 5μm);以醋酸盐缓冲液(取醋酸钠6.0g,加水4000ml溶解,加三乙胺800μl,四氢呋喃24ml,混匀,用2%醋酸溶液调pH值至7.2)为流动相A,以醋酸盐缓冲液-乙腈-甲醇(4∶7∶9)(取醋酸钠10.9g,加水800ml溶解,用2%醋酸溶液调pH值至7.2,加乙腈1400ml,甲醇1800ml,混匀)为流动相B,按下表进行梯度洗脱;检测波长为338nm(一级氨基酸)及262nm(二级氨基酸);柱温为35℃。
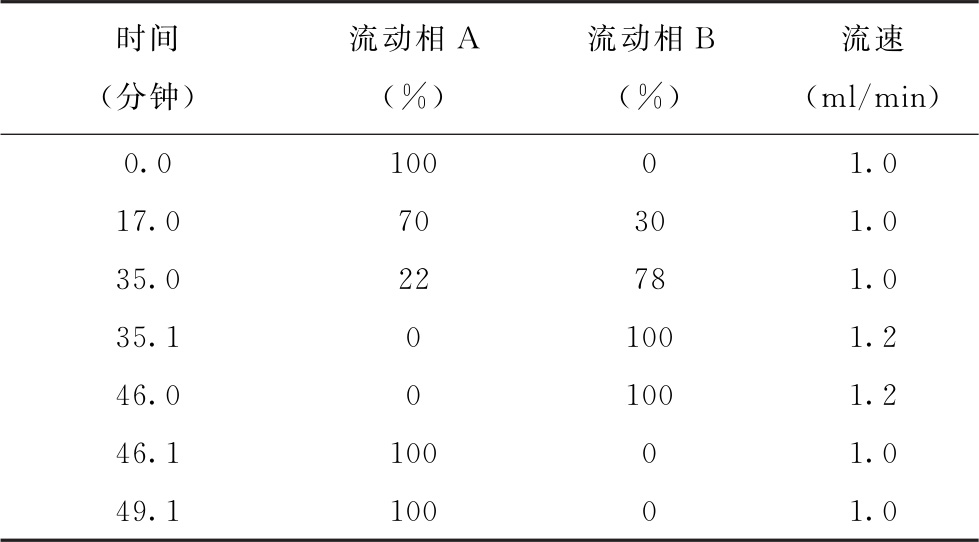
测定法 视供试品中待测氨基酸种类及其含量,取供试品、对照品及内标物(如正缬氨酸和肌氨酸)适量,用水或0.1mol/L盐酸溶液制成含总氨基酸浓度不大于5mg/ml的供试品溶液和与供试品溶液浓度相当的对照品溶液。精密量取供试品溶液10μl,置1.5ml塑料离心管中,精密加入0.4mol/L pH 10.2硼酸盐缓冲液(取硼酸24.7g,加水800ml溶解,用40%氢氧化钠溶液调pH值至10.2,然后加水稀释至1000ml)50μl,混匀,精密加入OPA溶液(取OPA 80mg,加0.4mol/L pH 10.2硼酸盐缓冲液7ml,加乙腈1ml, 3-巯基丙酸125μl,混匀)10μl,混匀,放置30秒,精密加入FMOC溶液(取FMOC 40mg,加乙腈8ml溶解)10μl,混匀,放置30秒,加水320μl,混匀,立即精密量取40μl,注入液相色谱仪,记录色谱图;另精密量取对照品溶液10μl,自“置1.5ml塑料离心管中”起同法测定。按内标法计算供试品溶液中各氨基酸的含量。
方法四 柱前DNFB衍生氨基酸测定法
原理 氨基酸与2,4-二硝基氟苯(DNFB)反应,生成有紫外响应的二硝基苯-氨基酸(DNP-氨基酸);DNP-氨基酸经反相高效液相色谱分离,用紫外检测器在360nm波长处检测,在一定的浓度范围(8~1000pmol)内,DNP-氨基酸吸光度与氨基酸浓度成正比。
特点 该法具有仪器配置要求不高、实验成本低的优点,但分离效果一般,适用于含氨基酸种类较少的样品分析。DNP-氨基酸衍生物溶液在室温下至少可稳定24小时;2,4-二硝基氟苯属易爆剧毒物质,有强致癌性,实验过程中应做好防护措施。
色谱条件 用十八烷基硅烷键合硅胶为填充剂(4.6mm×250mm, 5μm);以0.05mol/L醋酸钠溶液(取6.8g醋酸钠,加水800ml溶解,加二甲基甲酰胺10ml,用醋酸调pH值至6.4,用水稀释至1000ml)为流动相A,以乙腈-流动相A(1∶1)为流动相B,按下表进行梯度洗脱,流速为每分钟1ml;检测波长为360nm;柱温为40℃。
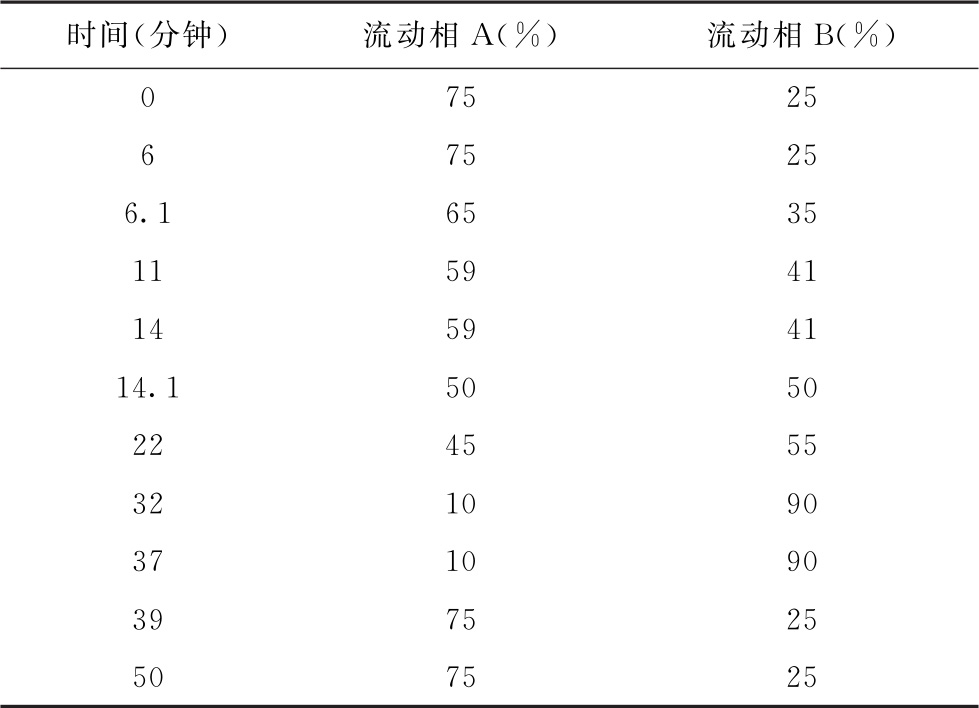
测定法 视供试品中待测氨基酸种类及其含量,取供试品、对照品适量,用水或0.1mol/L盐酸溶液制成含总氨基酸浓度不大于2.5mg/ml的供试品溶液和与供试品溶液浓度相当的对照品溶液。精密量取供试品溶液2ml,置50ml量瓶中,加0.5mol/L碳酸氢钠溶液2ml,加2,4-二硝基氟苯溶液(取2,4-二硝基氟苯1ml,用乙腈稀释至100ml)1ml,混匀,置60℃水浴中反应1小时,用pH 7.0磷酸盐缓冲液稀释至刻度,摇匀,精密量取20μl,注入液相色谱仪,记录色谱图;另精密量取对照品溶液2ml,自“置50ml量瓶中”起同法测定。按外标法计算供试品溶液中各氨基酸的含量。
方法五 柱后茚三酮衍生氨基酸锂离子交换系统测定法
原理 通过调节系统pH值及离子强度,采用锂离子交换系统,实现离子交换色谱柱对混合氨基酸的分离;经离子交换色谱分离的氨基酸与茚三酮反应,一级氨基酸生成紫色化合物,在570nm波长处有最大吸收。二级氨基酸(如脯氨酸)生成黄色化合物,在440nm波长处有最大吸收。在440nm和570nm波长处分别检测,在一定的浓度范围(20~500pmol)内,氨基酸衍生物的吸光度与氨基酸浓度成正比。
特点 该法使用氨基酸分析仪,具有自动化程度高、不易受基质干扰、重复性好等优点,适用于分析成分复杂样品。但色谱柱价格高、分析时间长、流动相制备复杂、部分氨基酸难于达到基线分离(如异亮氨酸和亮氨酸间的分离);采用温度梯度可以改善分离效果;尽量使用稀释液作为最后一步制备供试品溶液和对照品溶液的溶剂。
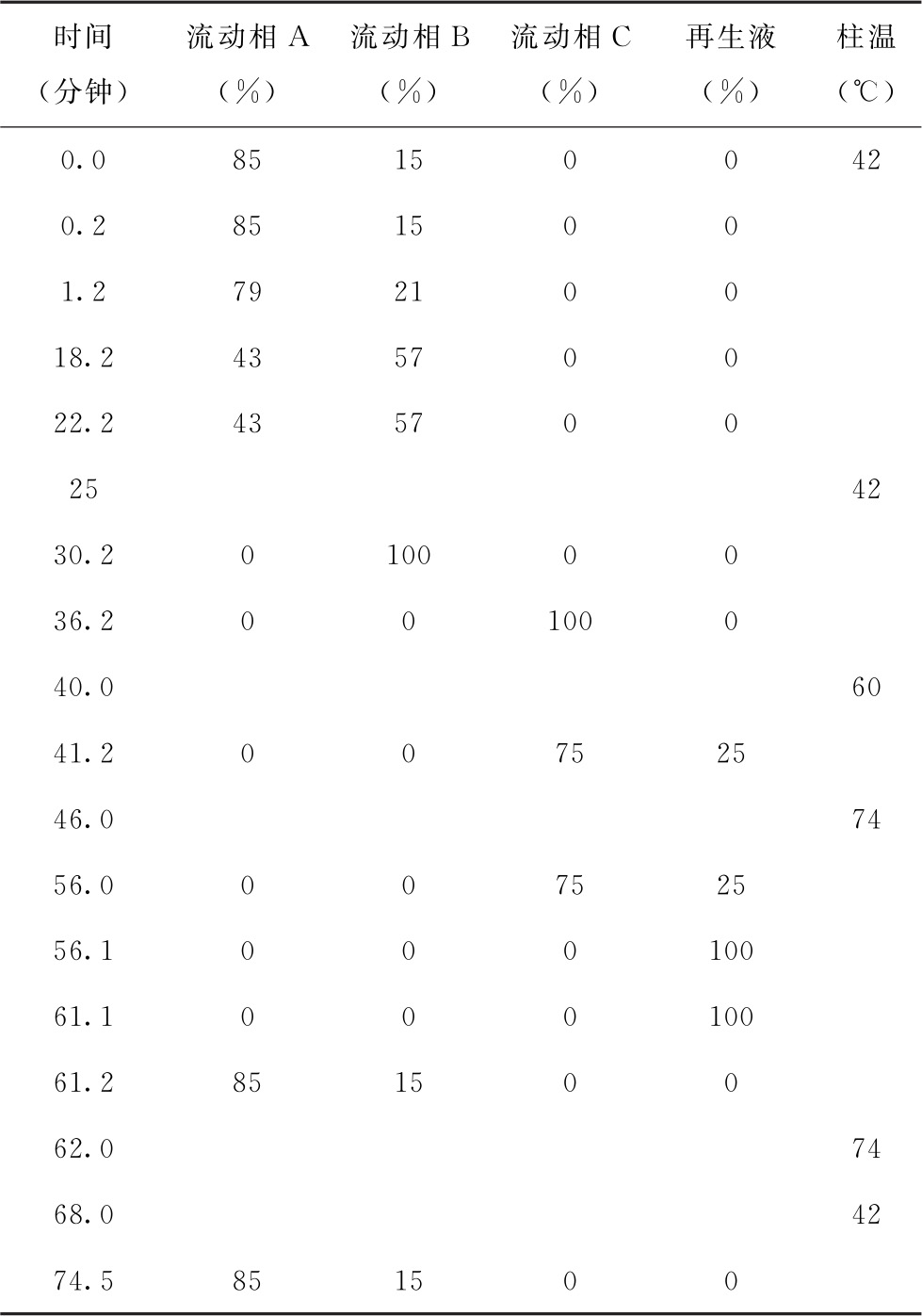
溶液配制 流动相A: 取氢氧化锂5.0g,枸橼酸16.4g,加水700ml溶解,加盐酸7.8ml,甲醇50ml,辛酸0.1ml,混匀,用盐酸或氢氧化锂溶液调节pH值至2.90±0.03,用水稀释定容至1L。流动相B: 取氢氧化锂8.4g,氯化锂4.2g,枸橼酸16.4g,加水700ml溶解,加盐酸8.6ml,辛酸0.1ml,混匀,用盐酸或氢氧化锂溶液调节pH值至4.20±0.05,用水稀释定容至1L。流动相C: 取氢氧化锂8.4g,氯化锂4.2g,枸橼酸10.9g,硼酸10.0g,加水700ml溶解,加盐酸3.3ml,辛酸0.1ml,混匀,用盐酸或氢氧化锂溶液调节pH值至8.00±0.05,用水稀释定容至1L。再生液: 取氢氧化锂21.0g,乙二胺四乙酸二钠0.2g,加水溶解,混匀,用水稀释定容至1L。衍生溶液: 取茚三酮20g,加甲醇600ml,苯酚2g,搅拌使溶解,加入醋酸钾钠缓冲液(取醋酸钠272.0g,醋酸钾196.0g,加入水约500ml,冰醋酸200ml,溶解,混匀,用醋酸溶液调pH值至5.55±0.05,用水稀释定容至1000ml)400ml,混匀,加入0.2g抗坏血酸(用少量甲醇溶解后加入),摇匀,即得。稀释液: 取氢氧化锂5.04g,枸橼酸17.5g,加水700ml溶解,加盐酸8.6ml,辛酸0.1ml,混匀,用盐酸或氢氧化锂溶液调节pH值至2.20±0.05,用水稀释定容至1L。
色谱条件 用锂离子型磺酸基强酸性阳离子交换树脂柱为填充剂(4.6mm×150mm);流动相流速为每分钟0.45ml,衍生溶液流速为每分钟0.25ml,按下表进行梯度洗脱和程序升温;反应器温度为130℃;检测波长为570nm(一级氨基酸)和440nm(二级氨基酸)。
测定法 视供试品中待测氨基酸种类及其含量,取供试品、对照品适量,用稀释液制成含总氨基酸浓度不大于1.5mg/ml的供试品溶液和与供试品溶液浓度相当的对照品溶液。精密量取氨基酸对照品溶液与供试品溶液50μl,分别注入氨基酸分析仪,记录色谱图;按外标法计算供试品溶液中各氨基酸的含量。
方法六 柱后茚三酮衍生氨基酸钠离子交换系统测定法
原理 通过调节系统pH值及离子强度,采用钠离子交换系统,实现离子交换色谱柱对混合氨基酸的分离,经离子交换色谱分离的氨基酸与茚三酮反应,一级氨基酸生成紫色化合物,在570nm波长处有最大吸收。二级氨基酸(如脯氨酸)生成黄色化合物,在440nm波长处有最大吸收。在440nm和570nm波长处分别检测,在一定的浓度范围(20~500pmol)内,氨基酸衍生物的吸光度与氨基酸浓度成正比。
特点 该法使用氨基酸分析仪,具有自动化程度高、不易受基质干扰、重复性好等优点,适用于分析比较简单的氨基酸混合物。
溶液配制 流动相A: 取枸橼酸钠11.8g、枸橼酸6.6g、苯酚0.5g、甲醇65ml、盐酸5.6ml、辛酸0.1ml,加水溶解并稀释至1000ml,用10%氢氧化钠溶液或盐酸调pH值至3.45±0.03。流动相B: 取枸橼酸钠19.6g、氢氧化钠3.1g、硼酸5.0g、辛酸0.1ml,加水溶解并稀释至1000ml,用10%氢氧化钠溶液或盐酸调pH值至10.86±0.05。再生液: 取氢氧化钠20.0g、乙二胺四乙酸二钠0.2g,加水溶解并稀释至1000ml。稀释液: 取枸橼酸钠11.8g、枸橼酸6.6g、苯酚2.0g、盐酸10.4ml、辛酸0.1ml,加水溶解并稀释至1000ml,用10%氢氧化钠溶液或盐酸调pH值至2.20±0.05。衍生溶液: 取茚三酮20g,加甲醇600ml,苯酚2g,搅拌使溶解,加入醋酸钾钠缓冲液(取醋酸钠272.0g,醋酸钾196.0g,加入水约500ml,冰醋酸200ml,使溶解,混匀,用醋酸溶液调pH值至5.55±0.05,加水稀释至1000ml)400ml,混匀,加入0.2g抗坏血酸(用少量甲醇溶解后加入),摇匀,即得。
色谱条件 用钠离子型磺酸基强酸性阳离子交换树脂为填充剂(4.6mm×150mm);流动相流速为每分钟0.45ml,衍生溶液流速为每分钟0.25ml,按下表进行梯度洗脱和程序升温;反应器温度为130℃;检测波长为570nm(一级氨基酸)和440nm(二级氨基酸)。
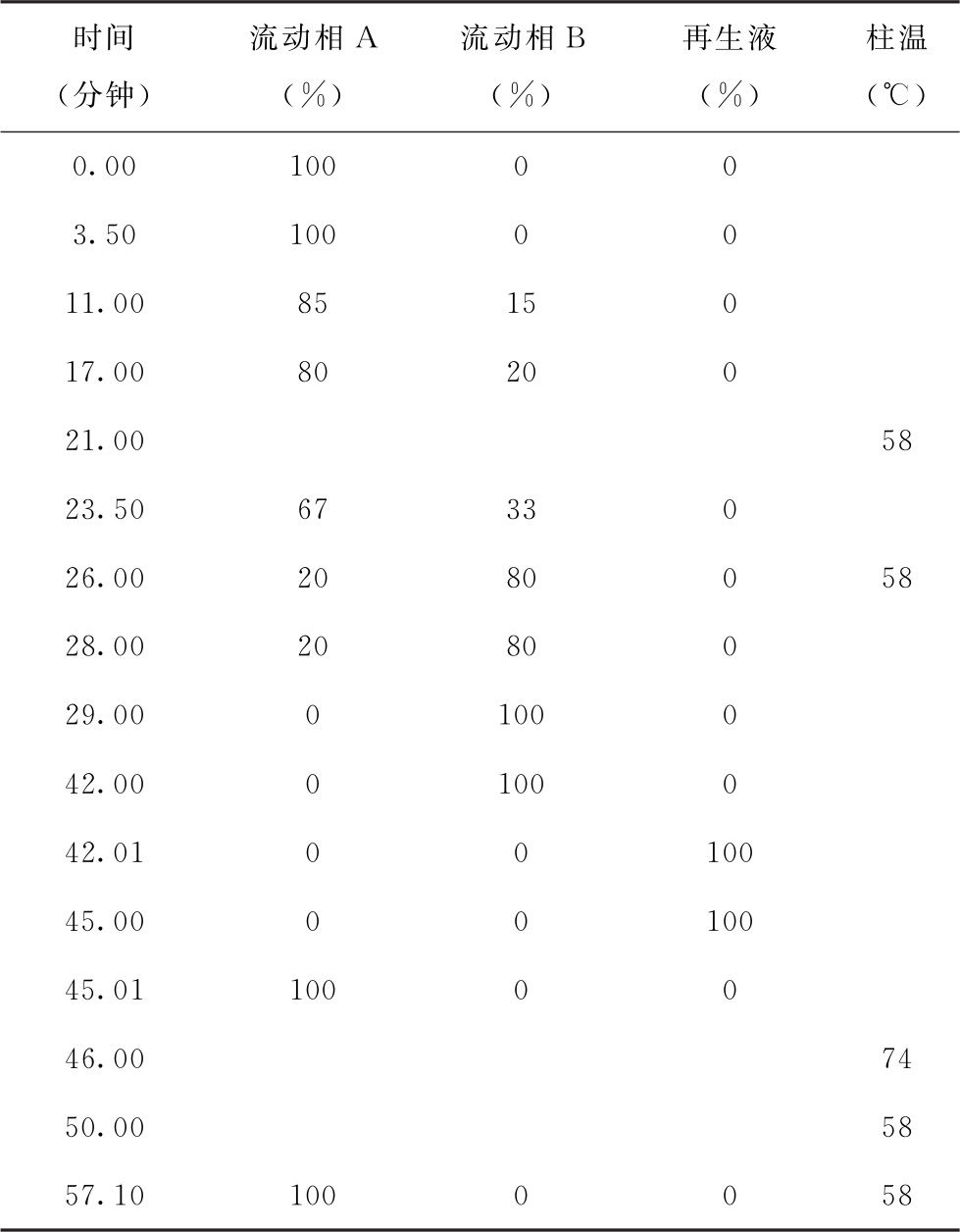
测定法 视供试品中待测氨基酸种类及其含量,取供试品适量,用稀释液制成含总氨基酸浓度不大于2.5mg/ml的供试品溶液和与供试品溶液浓度相当的对照品溶液。精密量取氨基酸对照品溶液与供试品溶液20μl,分别注入氨基酸分析仪,记录色谱图;按外标法计算供试品溶液中各氨基酸的含量。
4 数据处理
氨基酸分析的数据处理涉及氨基酸含量、蛋白质或多肽的氨基酸比值及含量的计算等。
4.1 氨基酸含量
采用适宜的氨基酸测定法测定,按外标法或内标法以峰面积计算样品中的各种氨基酸含量,如复方氨基酸注射液、药物制剂中游离的氨基酸等。
4.2 蛋白质或多肽含量及氨基酸比值
用氨基酸分析数据可测定已知分子量及氨基酸组成的蛋白质或多肽样品的含量。
在水解处理中稳定的氨基酸常被选择用于蛋白质或多肽的定量。在水解处理中稳定的氨基酸主要有门冬氨酸或门冬酰胺、谷氨酸或谷氨酰胺、丙氨酸、亮氨酸、苯丙氨酸、赖氨酸和精氨酸。可以根据蛋白质或多肽样品的氨基酸序列及不同的氨基酸测定方法调整用于定量分析的氨基酸种类。
采用适宜的氨基酸测定法测定蛋白质或多肽中的各种水解氨基酸含量,通过将稳定的每种氨基酸含量(nmol)分别除以蛋白质或多肽中所含的该氨基酸残基的理论个数,即可获得该蛋白质或多肽的含量。
由每种稳定的氨基酸含量计算该蛋白质或多肽的平均含量。通常舍去与平均值偏差大于5%的蛋白质或多肽含量值,并重新计算剩余各值的平均值。
蛋白质或多肽样品的氨基酸比值:将每种氨基酸的含量除以蛋白质或多肽平均含量,即得。
4.3 蛋白质或多肽含量及氨基酸残基数的预测
利用氨基酸分析数据可评估未知蛋白质或多肽样品中的蛋白质或多肽的含量。按下式计算蛋白质或多肽经水解后得到的每种氨基酸的含量(μg)。
每种氨基酸含量=mMW/1000
式中 m为样品中每种氨基酸的实测含量(nmol);
MW为每一种氨基酸的分子量与水的分子量之差。
在对蛋白质或多肽水解过程中部分和完全被破坏的氨基酸进行适当校正后,根据测得的每种氨基酸含量的总和,即为所测蛋白质或多肽的估算含量。
如果能得到未知蛋白质或多肽的分子量,就可以预测未知蛋白质或多肽的氨基酸组成。按下式计算未知蛋白质或多肽中每种氨基酸残基的数量。
每种氨基酸残基个数=m/(1000M/MWT)
式中 m为样品中每种氨基酸的实测含量(nmol);
M为蛋白质总含量(μg);
MWT是未知蛋白质或多肽的分子量。
(c)蒲标网 - 中国药典、药品标准、法规在线查询 ( 津ICP备15007510号 )