

3102 唾液酸测定法
第一法 间苯二酚显色法
本法系用酸水解方法将结合状态的唾液酸变成游离状态,游离状态的唾液酸与间苯二酚反应生成有色化合物,再用有机酸萃取后,测定唾液酸含量。
唾液酸对照品溶液(200μg/ml)的制备 精密称取唾液酸对照品10.52mg(1μg唾液酸相当于3.24nmol),置10ml量瓶中,加水溶解并稀释至刻度,混匀,即为唾液酸贮备液(1mg/ml),按一次使用量分装,-70℃贮存,有效期1年。仅可冻融1次。4℃保存,使用期为2周。精密量取唾液酸贮备液1ml,置5ml量瓶中,加水至刻度,即为每1ml含200μg的唾液酸对照品溶液,用前配制。
用于脑膜炎球菌多糖疫苗唾液酸含量测定时,同法制备浓度为400μg/ml的唾液酸对照品溶液贮备液(精密称取唾液酸40mg,置100ml量瓶中,用纯化水溶解并定容至刻度,混匀,即得)。精密量取唾液酸贮备液2.0ml,置10ml量瓶中,加水至刻度,即为每1ml含80μg的唾液酸对照品溶液,用前配制。
测定法 取供试品适量,加水稀释至蛋白质浓度为每1ml含0.2~0.4mg,作为供试品溶液。按下表取唾液酸对照品溶液、水及供试品溶液于10ml玻璃试管中,混匀,每管再加入间苯二酚-盐酸溶液(分别量取2%间苯二酚溶液2.5ml、0.1mol/L硫酸铜溶液62.5μl、25%盐酸溶液20ml,加水稀释至25ml,混匀。试验前4小时内配制)1ml,加盖,沸水煮沸30分钟(水浴面高于液面约2cm),取出置冰浴中3分钟(同时振摇)后,每管加乙酸丁酯-丁醇溶液(取4体积乙酸丁酯与1体积丁醇混匀,室温下保存,12小时内使用)2ml,充分混匀,室温放置10分钟,照紫外-可见分光光度法(通则0401),在波长580nm处测定吸光度。
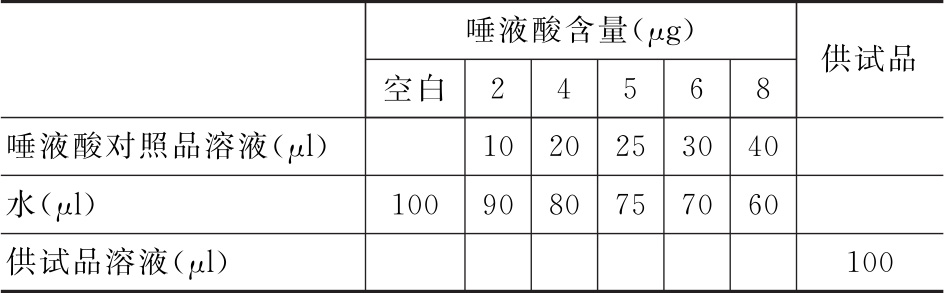
脑膜炎球菌疫苗唾液酸含量测定:取含唾液酸约40μg/ml的供试品溶液和纯水对照各2ml;另分别取唾液酸对照品(80μg/ml)0.1ml、0.2ml、0.4ml、0.8ml、1.6ml于各管中,补水至2.0ml为标准曲线各点。各管加2ml显色剂(0.1mol/L硫酸铜溶液0.5ml、4%间苯二酚溶液5ml、浓盐酸80ml,补水至100ml混匀。临用现配)摇匀,沸水浴15分钟后冰浴5~10分钟,每管加4ml有机相(正丁醇15ml,加乙酸丁酯定容至100ml),充分摇匀后置室温10分钟。以纯水对照为0点,于585nm测定吸光度,并作直线回归。(可按比例缩小供试品及各试剂体积)
以唾液酸对照品溶液的浓度对其相应的吸光度作直线回归(相关系数应不低于0.99),由直线回归方程计算5μg唾液酸的吸光度值,再按下式计算供试品唾液酸含量。

式中 A1为5μg唾液酸的吸光度;
A2为供试品的吸光度;
n为供试品稀释倍数;
P为供试品蛋白质含量,μg/μl;
W为1nmol促红素的量,相当于30.6μg。
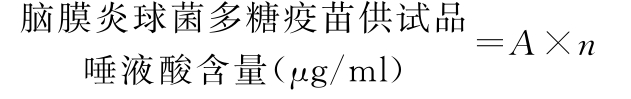
式中 A为供试品溶液吸光度相对于唾液酸对照品溶液的浓度,μg/ml;
n为供试品的稀释倍数。
第二法 超高效液相色谱法
本法系通过醋酸水解释放糖蛋白的唾液酸,再对释放的唾液酸进行标记,然后用超高效液相色谱(UPLC)对糖蛋白中的唾液酸进行测定。
照高效液相色谱法(通则0512)测定。
试剂 (1)醋酸溶液 取冰醋酸12g,加水至100ml,混匀。
(2)衍生溶液 避光操作。取水1.5ml、冰醋酸172μl和2-巯基乙醇112μl,混匀,加连二亚硫酸钠4.9mg,使溶解;再加4,5-亚甲二氧基-1,2-苯二胺二盐酸盐(DMB)3.5mg,加水200μl使充分溶解并混匀。
对照品溶液 取N-乙酰神经氨酸(Neu5Ac)约10mg,精密称定,置10ml量瓶中,加水溶解并稀释至刻度,作为贮备液(1);取N-羟乙酰神经氨酸(Neu5Gc)约10mg,精密称定,置250ml量瓶中,加水溶解并稀释至刻度,作为贮备液(2);精密量取贮备液(1)和贮备液(2)各4ml,置250ml量瓶中,用醋酸溶液稀释至刻度,作为混合对照品贮备液。
精密量取混合对照品贮备液0.4ml、0.8ml、2ml、4ml、8ml,分别置10ml量瓶中,用醋酸溶液稀释至刻度。精密量取以上溶液各200μl,置80℃孵育2~2.5小时,放冷。分别精密量取5μl,精密加入衍生溶液20μl,涡旋混匀,50℃避光孵育3小时,精密加水475μl终止反应,作为对照品溶液(1)~(5)。
供试品溶液 取供试品适量,置10kD超滤离心管中,不低于13 500转离心10分钟,弃去下层溶液。10kD超滤离心管中加水300μl,每分钟13 500转离心10分钟,弃去下层溶液,重复操作两次。取截留的上层溶液用适宜方法测定蛋白质含量,用醋酸溶液稀释至适宜浓度(含Neu5Ac约为10~20μmol/L),取200μl置80℃孵育2~2.5小时,放冷,精密量取5μl,加精密量取的衍生溶液20μl,涡旋混匀,50℃避光孵育3小时,精密加水475μl终止反应并混匀。
空白溶液 精密量取醋酸溶液200μl,自供试品溶液项下“置80℃孵育2~2.5小时”起,同法制备。
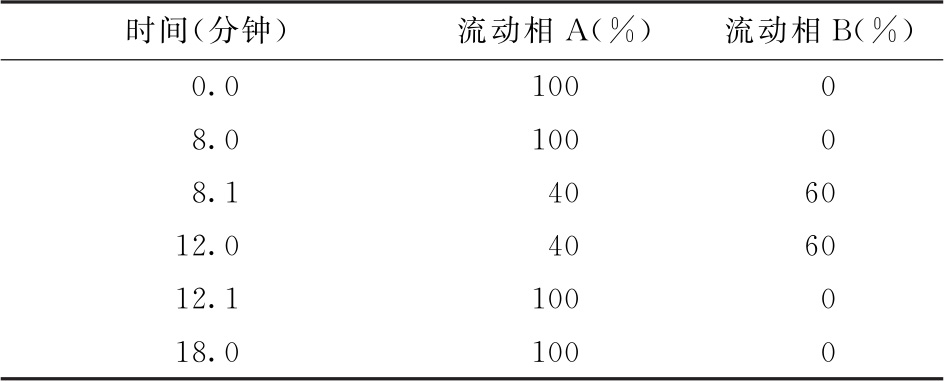
色谱条件 用十八烷基硅烷键合硅胶色谱柱(2.1mm×100mm, 1.7μm,或等效的色谱柱),柱温为30℃;以乙腈-甲醇-水(9∶7∶84)为流动相A,以乙腈为流动相B,按下表进行梯度洗脱,流速为每分钟0.25ml;荧光检测器,激发波长为373nm,发射波长为448nm;样品室温度为2~8℃,进样体积为5μl。
系统适用性要求 空白溶液色谱图中应无干扰峰。
对照品溶液(3)色谱图中Neu5Ac峰和Neu5Gc峰之间的分离度应不小于2.0,重复进样Neu5Ac和Neu5Gc峰面积的相对标准偏差(RSD)均应不大于5%(n=6);对照品溶液(1)色谱图中Neu5Gc峰的信噪比应不小于10。
以对照品溶液(1)~(5)中Neu5Ac和Neu5Gc浓度分别对其对应的峰面积计算线性回归方程,相关系数(r)均应不小于0.99。
测定法 精密量取空白溶液、对照品溶液(1)~(5)与供试品溶液,依序注入液相色谱仪,顺序为空白溶液(进样1针)、对照品溶液(3)(进样6针)、对照品溶液(1)~(5)、供试品溶液(1)、供试品溶液(2)……对照品溶液(3)(各进样1针),记录色谱图。
以对照品溶液(1)~(5)中Neu5Ac和Neu5Gc的浓度为横坐标,以其对应的峰面积为纵坐标,作线性回归方程。根据测得的供试品溶液峰面积,从线性方程分别计算Neu5Ac和Neu5Gc的浓度,按下式计算供试品中唾液酸(Neu5Ac或Neu5Gc)含量。1mg Neu5Ac相当于3.24μmol, 1mg Neu5Gc相当于3.08μmol。

式中 A为供试品溶液中Neu5Ac或Neu5Gc的浓度,μmol/L;
P为供试品溶液中蛋白质含量,mg/ml;
W为每1μmol蛋白质的量,相当于重量,mg;
n为供试品稀释倍数。
注意事项 (1)荧光检测器的增益可进行调节,以获得合适的信号响应强度。
(2)可采用优级纯试剂制备衍生溶液以避免产生干扰。该溶液可在-20℃条件暗处保存一年。衍生溶液也可采用经验证的商品化试剂盒。
(3)Neu5Ac和Neu5Gc均在0.04~40μmol/L浓度范围内,分别与其相应的峰面积呈良好线性。在线性范围内,可根据供试品中Neu5Ac和Neu5Gc的含量制备适宜浓度的对照品溶液(1)~(5)。
(4)如供试品中唾液酸含量较低,加水超滤换液后也可用4mol/L醋酸溶液进行稀释以保证供试品溶液中醋酸溶液的终浓度为2mol/L。
(5)唾液酸除常见的Neu5Ac和Neu5Gc外,还包括O-乙酰化的唾液酸(Neu5,7Ac2、Neu5Gc, 9Ac、Neu5,8Ac2、Neu5,9Ac2、Neu5,x,xAc3),详见图1。本法可同时分离检测7种唾液酸结构,详见图2。按面积归一化法,可计算供试品中各唾液酸占总唾液酸(7种唾液酸)的含量。若需要检测去O-乙酰化的Neu5Ac和Neu5Gc总量(包括5位氨基和4、7、8、9位的羟基发生取代),可在唾液酸释放之前进行去O-乙酰化处理(在供试品中加入10%体积的1mol/L氢氧化钠溶液,放置30分钟后,再加入10%体积的1mol/L盐酸溶液终止反应)。
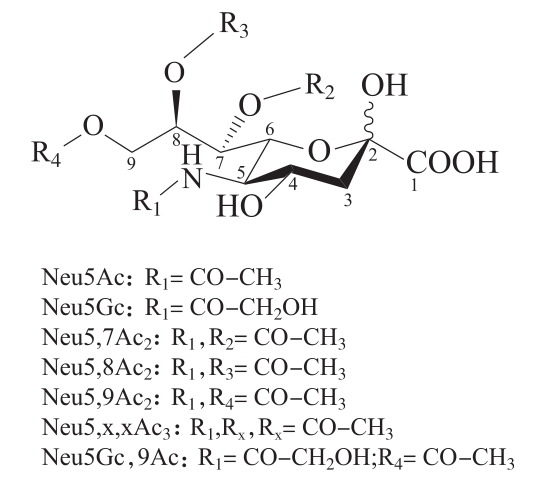
图1 唾液酸结构图
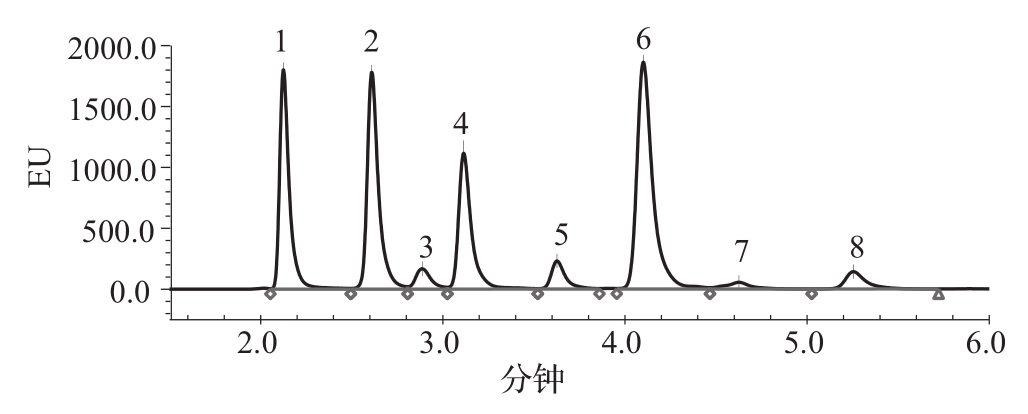
图2 唾液酸混合对照品(包含7种唾液酸结构)液相图谱
1.Neu5Gc 2.Neu5Ac 3.Neu5,7Ac2 4.Neu5Gc, 9Ac 5.Neu5,8Ac 6.Neu5,9Ac2 7.溶剂 8.Neu5,x,xAc3
第三法 高效液相色谱法
本法系通过醋酸水解释放糖蛋白的唾液酸,再对释放的唾液酸进行标记,然后用高效液相色谱(HPLC)对糖蛋白中的唾液酸进行测定。
照高效液相色谱法(通则0512)测定。
试剂、对照品溶液、供试品溶液、空白溶液、系统适用性要求、测定法和注意事项见第二法超高效液相色谱法。
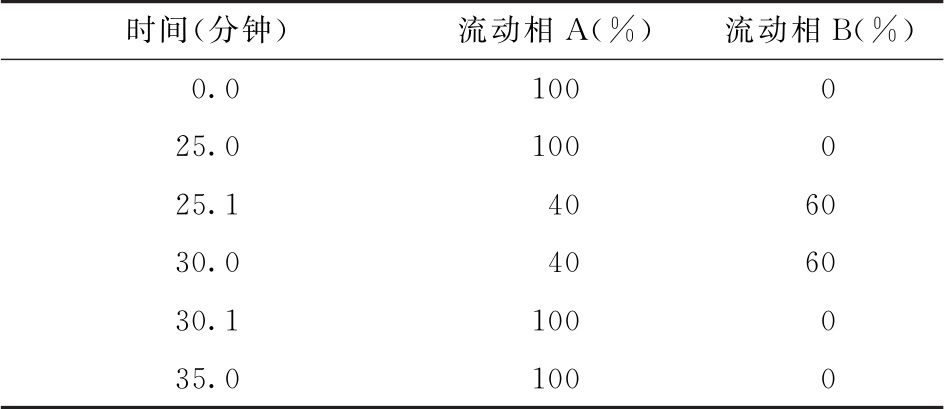
色谱条件 用十八烷基硅烷键合硅胶色谱柱(4.6mm×150mm, 3μm,或等效的色谱柱),柱温为30℃;以乙腈-甲醇-水(9∶7∶84)为流动相A,以乙腈为流动相B,按下表进行梯度洗脱,流速为每分钟0.5ml;荧光检测器,激发波长为373nm,发射波长为448nm;样品室温度为2~8℃,进样体积为25μl。
第四法 离子色谱法
本法系通过醋酸水解释放糖蛋白上的唾液酸,再采用离子交换色谱-脉冲安培检测器(HPAEC-PAD)对糖蛋白中的唾液酸进行测定。
照离子色谱法(通则0513)测定。
试剂 (1)样品缓冲液(20mmol/L醋酸钠溶液) 取无水醋酸钠164mg,加水80ml使溶解,用冰醋酸调节pH值至5.2,用水稀释至100ml,混匀。
(2)醋酸溶液 取冰醋酸12g,加水至100ml,混匀。
对照品溶液 取N-乙酰神经氨酸(Neu5Ac)约10mg,精密称定,置10ml量瓶中,加水溶解并稀释至刻度,作为贮备液(1);取N-羟乙酰神经氨酸(Neu5Gc)约10mg,精密称定,置250ml量瓶中,加水溶解并稀释至刻度,作为贮备液(2);精密量取贮备液(1)和贮备液(2)各4ml,置250ml量瓶中,用水稀释至刻度,作为混合对照品贮备液。
精密量取混合对照品贮备液5μl、10μl、25μl、50μl、100μl,分别置1.5ml离心管中,冷冻离心干燥,精密加入醋酸溶液100μl,置80℃孵育2~2.5小时,放冷,涡旋混匀,冷冻离心干燥,精密加入水300μl复溶,再加入精密量取的样品缓冲液200μl,涡旋混匀,转移至10kD超滤离心管中,在2~8℃条件下,每分钟4000转离心15分钟,取下层溶液,作为对照品溶液(1)~(5)。
供试品溶液 取供试品适量,用醋酸溶液稀释至适宜浓度(含Neu5Ac约为10~20μmol/L),精密量取100μl,置80℃孵育2~2.5小时,放冷,涡旋混匀,冷冻离心干燥,精密加入水300μl复溶,再加入精密量取的样品缓冲液200μl,涡旋混匀,转移至10kD超滤离心管中,在2~8℃条件下,每分钟4000转离心15分钟,取下层溶液。
空白溶液 精密量取醋酸溶液100μl,自供试品溶液项下“置80℃孵育2~2.5小时”起,同法制备。
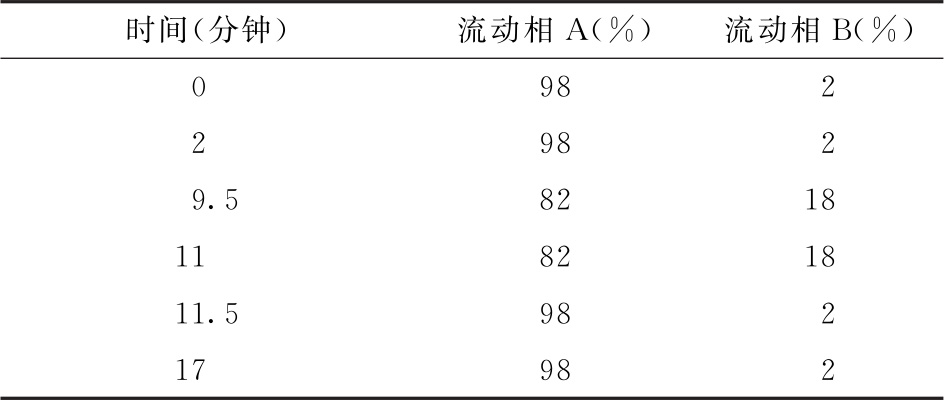
色谱条件 用糖分析柱(3mm×150mm,或等效的色谱柱),保护柱(3mm×30mm,或等效的色谱柱);柱温为30℃;以0.1mol/L氢氧化钠溶液为流动相A,以含1mol/L醋酸钠和0.1mol/L氢氧化钠的水溶液为流动相B,按下表进行梯度洗脱;流速为每分钟0.5ml;样品室温度为2~8℃;进样体积为25μl。
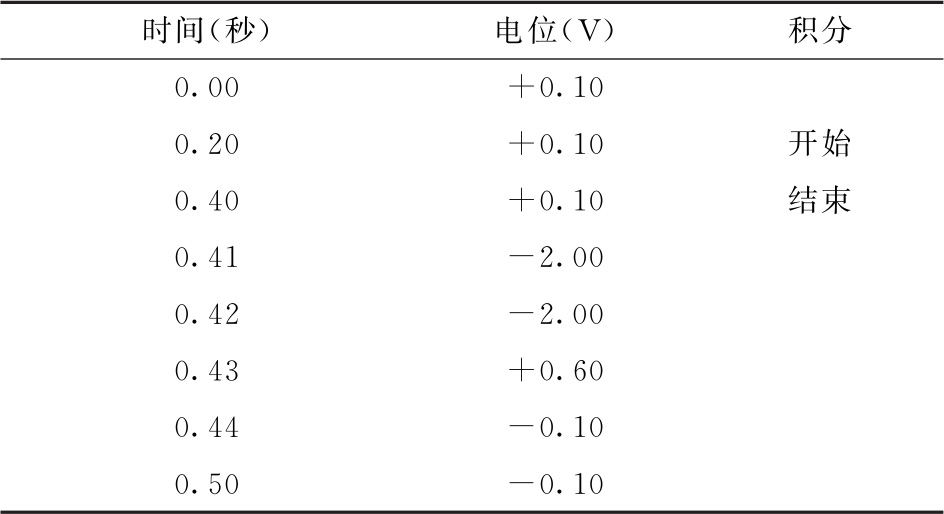
检测器为脉冲安培检测器(PAD),Au工作电极(推荐使用1mm直径)、Ag/AgCl参比电极,四电位检测波形(电位见下表)进行检测。
系统适用性要求 空白溶液色谱图中应无干扰峰。
对照品溶液(3)色谱图中Neu5Ac峰和Neu5Gc峰之间的分离度应不小于2.0,重复进样Neu5Ac和Neu5Gc峰面积的相对标准偏差(RSD)均应不大于5%(n=6);对照品溶液(1)色谱图中Neu5Gc峰的信噪比应不小于10。
以对照品溶液(1)~(5)中Neu5Ac和Neu5Gc浓度分别与其对应的峰面积计算线性回归方程,相关系数(r)均应不小于0.99。
序列后对照品溶液(3)色谱图中Neu5Ac和Neu5Gc的峰面积,应为序列前对照品溶液(3)重复进样6针平均峰面积的90%~110%。
测定法 精密量取空白溶液、对照品溶液(1)~(5)与供试品溶液,依序注入离子色谱仪,顺序为空白溶液(进样1针)、对照品溶液(3)(进样6针)、对照品溶液(1)~(5)、供试品溶液(1)、供试品溶液(2)……对照品溶液(3)(各进样1针),记录色谱图。
以对照品溶液(1)~(5)中Neu5Ac和Neu5Gc的浓度为横坐标,以其对应的峰面积为纵坐标,作线性回归方程。根据测得的供试品溶液峰面积,从线性方程分别计算Neu5Ac和Neu5Gc的浓度,再按下式计算供试品中唾液酸(Neu5Ac或Neu5Gc)含量。1mg Neu5Ac相当于3.24μmol, 1mg Neu5Gc相当于3.08μmol。

式中 A为供试品溶液中Neu5Ac或Neu5Gc的浓度,μmol/L;
P为供试品溶液中蛋白质含量,mg/ml;
W为每1μmol蛋白质的量,相当于重量,mg;
n为供试品稀释倍数。
注意事项 (1)样品中蛋白质的存在可能会降低PAD的响应,为保证检测结果的准确性,每进样10针供试品溶液后应进样1针对照品溶液(3)。
(2)为减小PAD响应降低的影响,可使用3-脱氧-D-甘油-D-半乳壬酮糖(KDN)作为内标。具体操作为:取0.1mmol/L KDN溶液作为内标溶液,在对照品溶液(1)~(5)与供试品溶液制备的最后稀释步骤中,分别加内标溶液15μl。以加内标的直线回归方程分别计算供试品溶液中Neu5Ac和Neu5Gc的含量。
(3)Neu5Ac和Neu5Gc均在0.01~10μmol/L浓度范围内,分别与其相应的峰面积呈良好线性。在线性范围内,可根据供试品中Neu5Ac和Neu5Gc的含量制备适宜浓度的对照品溶液(1)~(5)。
(4)不同品牌检测器可能存在差异,可对检测器参数(电位电压等)进行适当调整,以获得合适的信号响应强度。
(5)离子色谱仪品牌不同,色谱柱的品牌/批号不同,对照品溶液的色谱图与参考图谱(图3)在峰型上可能略有差异。可根据色谱柱说明书对色谱条件进行适当调整。
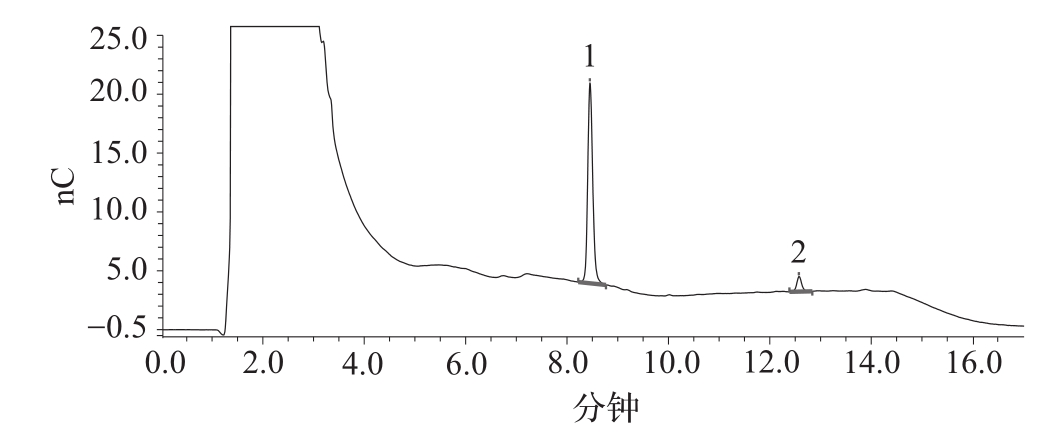
图3 对照品溶液离子色谱图谱
1.Neu5Ac 2.Neu5Gc
(c)蒲标网 - 中国药典、药品标准、法规在线查询 ( 津ICP备15007510号 )