
药典目录
9303 色素测定法指导原则
来源:四部 分类:通则 页码:4049303 色素测定法指导原则 色素一般分为天然色素和人工合成色素。药品生产中色素的使用应符合国家药品管理部门的有关规定。中药(中药材和中药饮片,下同)不应使用色素。本指导原则为药品(中药、化药、辅料)中色素的检测提供指导。 基本原则 辅料、化药、中药等基质情况各不相同,如需检测色素时应注意基质的影响,对不同基质应分别采用针对性的前处理方法。对于中药应对与其性状颜色相近的色素进行筛选,并准确地定性、定量。 基本内容 一、定性测定方法 常用的定性测定方法包括薄层色谱法、高效液相色谱法、高效液相色谱-质谱联用法等。 1.薄层色谱法 (1)对照品溶液的制备 应根据色素的理化性质,保持色素良好的溶解性和稳定性为原则,选择合适的溶剂并配制成合适的浓度。多种色素配制混合对照品溶液时,应注意合理分组,避免色素分解或相互发生反应。 如苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ、苏丹红G、分散红9、808猩红等脂溶性色素,可选用乙腈等作为溶剂。孔雀石绿、新品红、金胺O、罗丹明B、金橙Ⅰ、酸性黑Ⅰ、亮蓝G、亮蓝、羊毛绿、柠檬黄、日落黄、亮黄、金橙Ⅱ、酸性橙10、赤藓红、酸性红73、苋菜红、酸性红18、诱惑红、曙红等水溶性色素,可选用水作为溶剂。 (2)供试品溶液的制备 应考虑测定对象的基质情况和测定色素的理化特点,参考对照品溶液配制溶剂选用合适的溶剂,采用快速、简单、高效的前处理技术进行提取,必要时可进行净化处理,避免基质干扰。 如测定孔雀石绿、新品红、金胺O、罗丹明B等偏碱性水溶性色素,可采用0.1%甲酸甲醇溶液提取;测定金橙Ⅱ、赤藓红、酸性红73、苋菜红等偏酸性水溶性色素,可采用0.1%甲酸甲醇溶液提取。通过超声处理30分钟,离心后的上清液作为供试品溶液。 (3)薄层板和展开剂 可根据测定色素的数量和展开后的色谱效果,选择合适的普通或高效薄层板。应根据色素展开后,合理的Rf值和与相邻斑点的分离度,选择合适的展开剂。 如苏丹红I等脂溶性色素,采用环己烷-乙酸乙酯-氨水(80:20:1)的上层溶液为展开剂;孔雀石绿、金橙Ⅱ等水溶性色素,采用以乙酸乙酯-乙醇-水-氨水(6:4:2:0.5)的上层溶液为展开剂。 (4)检视方法 大多数色素可在日光下检视,必要时可在紫外光下进行检视。 (5)注意事项 在色素量较低或基质较为复杂的情况下,对薄层色谱法的定性,检测结果应注意排除假阴性或假阳性。 2.高效液相色谱法 (1)色谱条件与系统适用性试验 应根据色素的理化性质选择适宜的固定相和流动相,多种色素测定时可适当采用流动相梯度洗脱,达到良好分离效果。固定相常用十八烷基硅烷键合硅胶为填充剂,可根据情况选择小粒径或柱长较长的色谱柱以提高分离度。 如测定苏丹红I等脂溶性色素时,以甲醇为流动相A,0.1%甲酸溶液为流动相B,以下列梯度洗脱:0~12分钟,A:80%→100%;12~20分钟,A:100%。测定孔雀石绿等偏碱性水溶性色素时,以下列梯度洗脱:0~27分钟,A:40%→72%;27~27.1分钟,A:72%→95%;27.1~32分钟,A:95%。测定金橙Ⅱ等偏酸性水溶性色素时,以甲醇为流动相A,20mmol/L醋酸铵为流动相B,以下列梯度洗脱:0~7分钟,A:5%→45%;7~17分钟,A:45%→50%;17~20分钟,A:50%→65%;20~30分钟,A:65%→95%;30~32分钟,A:95%。 应根据各色素在紫外-可见光区的最大吸收波长选择适合的检测波长。如黄、橙、红、蓝绿色素系列建议分别选在400nm、440nm、520nm、610nm波长附近作为检测波长。 为增加定性结果的可靠性,应根据色素具体情况同时采集对照品与供试品不同波段的紫外-可见吸收光谱比较其是否一致。 (2)对照品溶液的制备 应采用适宜的溶剂配制合适的浓度,注意色素的溶解性和稳定性。可参照本指导原则1.薄层色谱法项下。 (3)供试品溶液的制备 应采用适宜的溶剂,采用快速、简单、高效的前处理技术进行提取;需尽量减少对色谱柱污染和复杂基质的影响,一般情况下需进一步净化,净化时应考虑合适的净化填料和洗脱流程,注意避免待测色素的损失。 如测定孔雀石绿等偏碱性水溶性色素,可对0.1%甲酸甲醇溶液的供试品提取溶液,采用聚酰胺填料净化,收集甲醇-0.1%甲酸溶液(3:2)混合溶液的洗脱液作为供试品溶液。测定金橙Ⅱ、赤藓红、酸性红73、苋菜红等偏酸性水溶性色素,可对供试品的甲醇-0.1%甲酸溶液(3:2)混合溶液提取液,采用聚酰胺填料净化,收集甲醇-氨水-水(7:2:1)洗脱液,调节pH值至弱酸性作为供试品溶液。 (4)结果判断 供试品色谱中,出现与相应对照品保留时间相同的色谱峰,并且其紫外-可见吸收光谱与对照品光谱相同时,可基本判断检出相应的色素。 (5)注意事项 对于复杂基质,应尽量在每针进样后以高比例有机相冲洗色谱柱,在色谱柱前加预柱或保护柱,以延长色谱柱寿命。 应注意本法测定时出现的假阳性情况或色谱图有干扰时,可通过其他适宜的方法予以确认。 在本方法未检出的情况下,也应注意假阴性情况,结合方法检测限,综合判断,必要情况下应采用更为灵敏的方法进行检测。 3.离效液相色谱-质谱联用法 (1)色谱条件与系统适用性试验 应根据色素的理化性质选择适宜的固定相和流动相,多种色素测定时可适当采用流动相梯度洗脱,特别应注意同分异构体的色素应达到良好分离效果。固定相常采用十八烷基硅烷键合硅胶为填充剂。 流动相组成的选择应注意色素在质谱中的采集模式,以提高离子化效率。如正离子模式下,可以乙腈-0.1%甲酸溶液(含20mmol/L醋酸铵)系统作为流动相;负离子模式下,可以乙腈-2mmol/L醋酸铵溶液系统作为流动相。 应根据仪器的具体情况,选择各色素最佳的离子采集模式,并对色素的质谱检测参数进行优化达到最佳。采用三重串联四极杆质谱作为检测器时,应分别对每种色素选择多对特征性的离子对通道,并针对性优化设定最佳碰撞电压(CE)等。见表1,表2。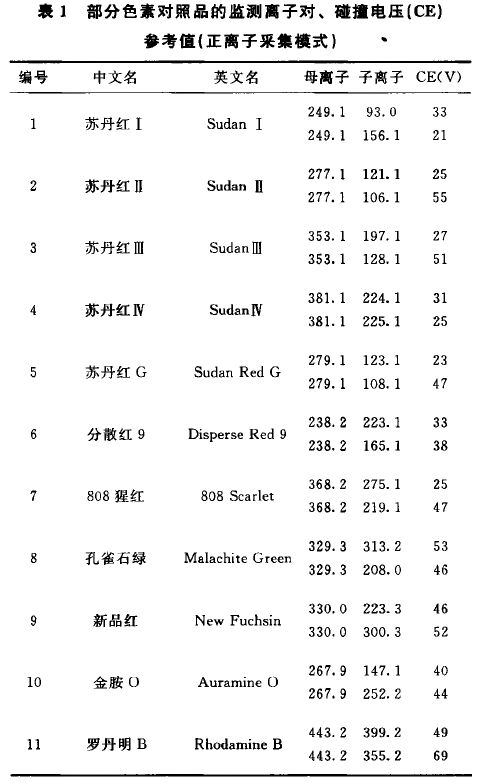
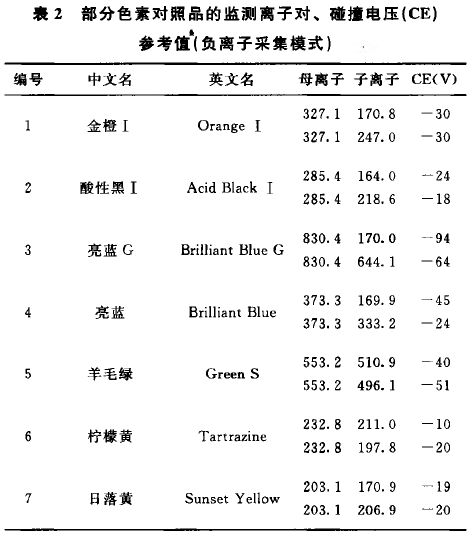
(2)对照品溶液的制备 可参考高效液相色谱法项下,适当稀释至合适浓度,作为对照品溶液。 (3)供试品溶液的制备 可参考高效液相色谱法项下,适当稀释至合适浓度,作为供试品溶液。 (4)结果判断 供试品色谱中如检出与对照品保留时间相同的色谱峰,并且所选择的多对子离子的质荷比一致,供试品溶液的定性离子相对丰度比与浓度相当对照品溶液的定性离子相对丰度比进行比较时,相对偏差不超过下列规定的范围,则可判定样品中存在该组分:相对比例>50%,允许±20%偏差;相对比例20%?50%,允许±25%偏差;相对比例10%?20%,允许±30%偏差;相对比例<10%,允许±50%偏差。 (5)注意事项 色谱-质谱联用方法作为定性确证方法,可采用多种不同原理的质谱作为检测技术,但均应保证结果的准确可靠。 应注意色素适宜的进样浓度,避免交叉污染或对系统造成残留污染,注意采用空白试剂等进行过程质量控制。 在未检出的情况下,应考虑方法检测限,综合判断。 二、含量测定方法 常用的含量测定方法包括高效液相色谱法、高效液相色谱-质谱联用法等。在经色谱-质谱联用法对定性结果确认的前提下,可采用高效液相色谱法进行定量。建立含量测定方法时应符合药品质量标准分析方法验证指导原则(通则9101)。 1.高效液相色谱法 (1)色谱条件与系统适用性试验 可参考本指导原则一、定性测定方法中2.高效液相色谱法项下有关内容。 (2)计算方法 可根据情况采用外标一点法、标准曲线法、内标法等准确测定计算待测色素的含量。 (3)对照品溶液的制备 应将对照品溶液配制至合适的浓度,并且与供试品中待测色素的浓度相近。或配制不同浓度水平的对照品标准曲线工作溶液。 (4)供试品溶液的制备 可采用适宜的提取手段多次提取以尽可能达到将色素提取完全,净化过程应注意避免色素损失。对中药取样时,应注意取样的均匀性和代表性,样品适当粉碎但不宜过细,以减少基质成分对测定的干扰。 2.高效液相色谱-质谱联用法 (1)色谱条件与系统适用性试验 可参考本指导原则一、定性测定方法中3.高效液相色谱-质谱联用法项下有关内容。 (2)计算方法、对照品溶液的制备、供试品溶液的制备 相关要求均可参考本部分1.高效液相色谱法相应内容。 (3)注意事项 如出现基质效应可采用空白基质溶液(即不含待测色素的同种样品按供试品溶液制备方法制得的溶液)配制标准曲线的方法予以消除。 亮蓝G、柠檬黄、日落黄、苋菜红和酸性红18等部分色素的质谱响应可能不高,标准曲线范围可根据仪器响应适当收窄。