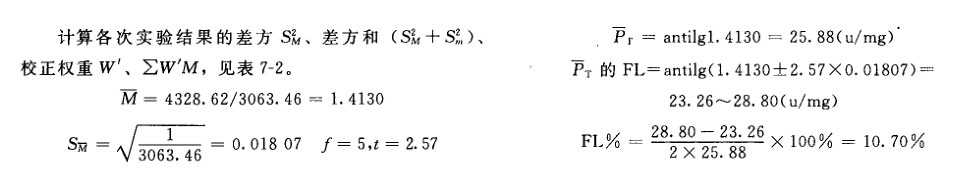药典目录
1431 生物检定统计法
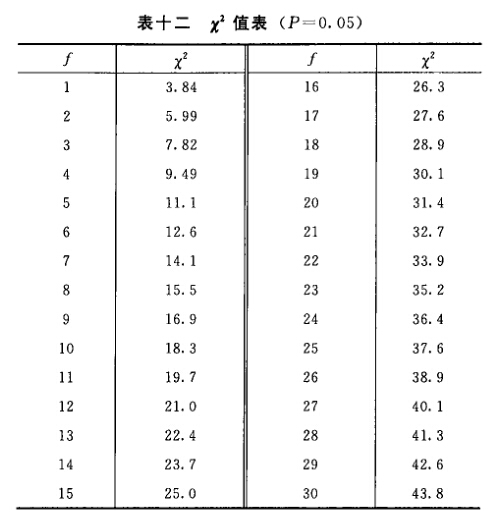
来源:三部 分类:三部通则 页码:5911431 生物检定统计法 —、总则 生物检定法是利用生物体包括整体动物、离体组织、器官、细胞和微生物等评估药物生物活性的一种方法。它以药物的药理作用为基础,以生物统计为工具,运用特定的实验设计在一定条件下比较供试品和相当的标准品或对照品所产生的特定反应,通过等反应剂量间比例的运算或限值剂量引起的生物反应程度,从而测定供试品的效价、生物活性或杂质引起的毒性。 生物检定统计法主要叙述应用生物检定时必须注意的基本原则、一般要求、实验设计及统计方法。有关品种用生物检定的具体实验条件和要求,必须按照该品种生物检定法项下的规定。 生物检定标准品 凡中国药典规定用生物检定的品种都有它的生物检定标准品(S)。S都有标示效价,以效价单位(u)表示,其含义和相应的国际标准品的效价单位一致。 供试品 供试品(T)或(U)是供检定其效价的样品,它的活性组分应与标准品基本相同。 AT或AU是T或U的标示量或估计效价。 等反应剂量对比 生物检定是将T和其S在相同的实验条件下同时对生物体或其离体器官组织等的作用进行比较,通过对比,计算出它们的等反应剂量比值(R),以测得T的效价PT。 R是S和T等反应剂量(ds、dT)的比值,即 R=ds/dT。 M是S和T的对数等反应剂量(xs、xT)之差,即M=lgds-lgdT=xs-xT。R=antilgM。 PT是通过检定测得T的效价含量,称T的测得效价,是将效价比值(R)用T的标示量或估计效价AT校正之后而得,即 PT=AT.R或PT=AT.antilgM。 检定时,S按标示效价计算剂量,T按标示量或估计效价(AT)计算剂量,注意调节T的剂量或调整其标示量或估计效价,使S和T的相应剂量组所致的反应程度相近。 生物变异的控制 生物检定具有一定的实验误差,其主要来源是生物变异性。因此生物检定必须注意控制生物变异或减少生物变异本身,或用适宜的实验设计来减小生物变异对实验结果的影响,以减小实验误差。控制生物变异必须注意以下几点。 (1)生物来源、饲养或培养条件必须均一。 (2)对影响实验误差的条件和因子,在实验设计时应尽可能作为因级限制,将选取的因级随机分配至各组。例如体重、性别、窝别、双碟和给药次序等都是因子,不同体重是体重因子的级,雌性、雄性是性别因子的级,不同窝的动物是窝别因子的级,不同双碟是碟间因子的级,给药先后是次序因子的级等。按程度划分的级(如动物体重),在选级时,应选动物较多的邻近几级,不要间隔跳越选级。 (3)按实验设计类型的要求将限制的因级分组时,也必须严格遵守随机的原则。 误差项 指从实验结果的总变异中分去不同剂量及不同因级对变异的影响后,剩余的变异成分,用方差(S2)表示。对于因实验设计类型的限制无法分离的变异成分,或估计某种因级对变异的影响小,可不予分离者,都并入S2 。但剂间变异必须分离。 误差项的大小影响标准误SM和可信限(FL)。 不同的检定方法和实验设计类型,分别按有关的公式计算S2。 可靠性测验 平行线检定要求在实验所用的剂量范围内,对数剂量的反应(或反应的函数)呈直线关系,供试品和标准品的直线应平行。可靠性测验即验证供试品和标准品的对数剂量反应关系是否显著偏离平行偏离直线,对不是显著偏离平行偏离直线(在一定的概率水平下)的实验结果,认为可靠性成立,方可按有关公式计算供试品的效价和可信限。 可信限和可信限率 可信限(FL)标志检定结果的精密度。M的可信限是M的标准误SM和t值的乘积(t.SM),用95%的概率水平。M+t.SM是可信限的高限;M-t.SM是可信限的低限。用其反对数计算得R和PT的可信限低限及高限,是在95%的概率水平下从样品的检定结果估计其真实结果的所在范围。 R或PT的可信限率(FL%)是用R或PT的可信限计算而得。效价的可信限率为可信限的高限与低限之差除以2倍平均数(或效价)后的百分率。计算可信限的t值是根据S2的自由度(f)查t值表而得。 t值与f的关系见表一。
各品种的检定方法项下都有其可信限率的规定,如果检定结果不符合规定,可缩小动物体重范围或年龄范围,或调整对供试品的估计效价或调节剂量,重复实验以减小可信限率。 对同批供试品重复试验所得n次实验结果(包括FL%超过规定的结果),可按实验结果的合并计算法算得PT的均值及其FL%作为检定结果。 二、直接测定法 直接测得药物对各个动物最小效量或最小致死量的检定方法。如洋地黄及其制剂的效价测定。

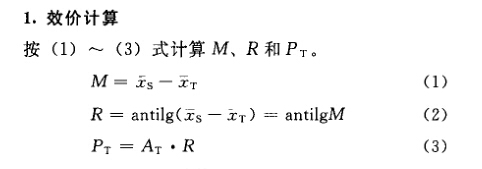


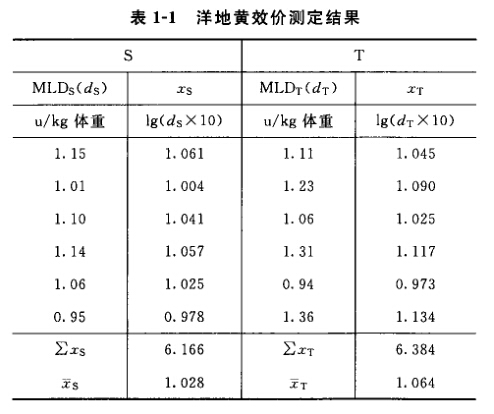
例1 直接测定法 洋地黄效价测定——鸽最小致死量(MLD)法 S为洋地黄标准品,按标示效价配成1.0u/ml的酊剂,临试验前稀释25倍。 T为洋地黄叶粉,估计效价AT=10u/g,配成1.0u/ml的酊剂,临试验前配成稀释液(1→25)。测定结果见表1-1。
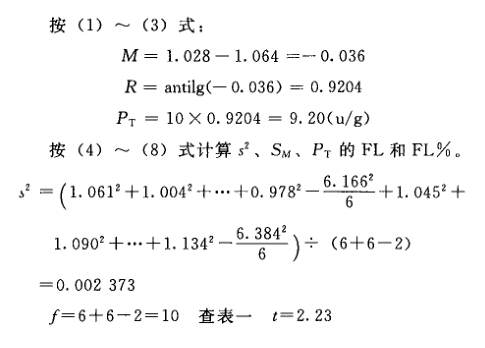
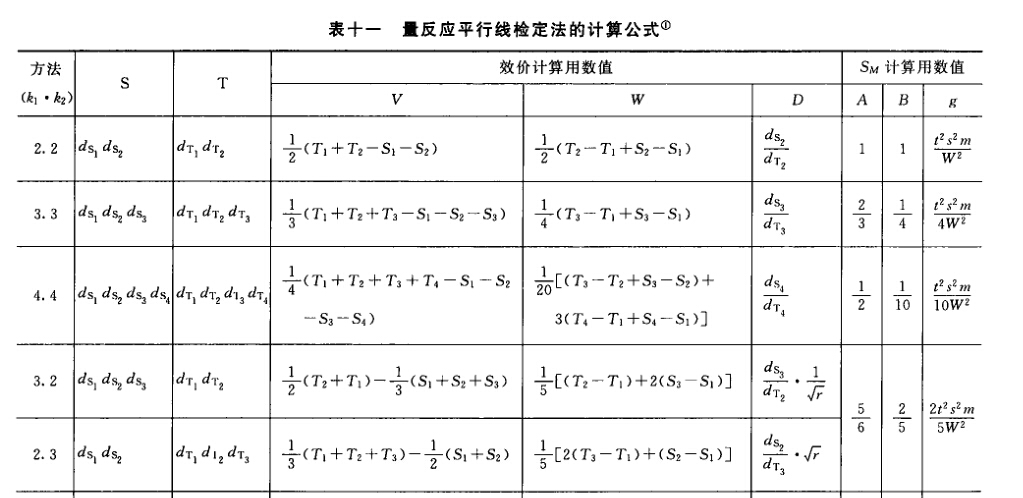
三、量反应平行线测定法 药物对生物体所引起的反应随着药物剂量的增加产生的量变可以测量者,称量反应。量反应检定用平行线测定法,要求在一定剂量范围内,S和T的对数剂量x和反应或反应的特定函数y呈直线关系,当S和T的活性组分基本相同时,两直线平行。 本版药典量反应检定主要用(2.2)法、(3.3)法或(2.2.2)法、(3.3.3)法,即S、T(或U)各用2个剂量组或3个剂量组,统称(k.k)法或(k.k.k)法;如果S和T的剂量组数不相等,则称(k. k')法;前面的k代表S的剂量组数,后面的k或k'代表T的剂量组数。一般都是按(k.k)法实验设计,当S或T的端剂量所致的反应未达阈值,或趋于极限,去除此端剂量后,对数剂量和反应的直线关系成立,这就形成了(k.k')法。例如(3.3)法设计就可能形成(2.3)法或(3.2)法等。因此,(k.k')法中的k只可能比k'多一组或少一组剂量。(k.k')法的计算结果可供重复试验时调节剂量或调整供试品估计效价时参考。无论是(k.k)法、(k.k')法或(k.k.k)法,都以K代表S和T的剂量组数之和,故K=k+k或K=k+k'或K=k+k+k。 本版药典平行线测定法的计算都用简算法,因此对各种(k.k)法要求: (1)S和T相邻高低剂量组的比值(r)要相等,一般r用(1:0.8)~(1:0.5),lgr=I (2)各剂量组的反应个数(m)应相等。 1.平行线测定的实验设计类型 根据不同的检定方法可加以限制的因级数采用不同的实验设计类型。本版药典主要用下面三种实验设计类型。 (1)随机设计 剂量组内不加因级限制,有关因子的各级随机分配到各剂量组。本设计类型的实验结果只能分离不同剂量(剂间)所致变异,如绒促性素的生物检定。 (2)随机区组设计 将实验动物或实验对象分成区组,一个区组可以是一窝动物、一只双碟或一次实验。在剂量组内的各行间加以区组间(如窝间、碟间、实验次序间)的因级限制。随机区组设计要求每一区组的容量(如每一窝动物的受试动物只数、每一只双碟能容纳的小杯数等)必须和剂量组数相同,这样可以使每一窝动物或每一只双碟都能接受到各个不同的剂量。因此随机区组设计除了从总变异中分离剂间变异之外,还可以分离区组间变异,减小实验误差。例如抗生素杯碟法效价测定。 (3)交叉设计 同一动物可以分两次进行实验者适合用交叉设计。交叉设计是将动物分组,每组可以是一只动物,也可以是几只动物,但各组的动物只数应相等。标准品(S)和供试品(T)对比时,一组动物在第一次试验时接受S的一个剂量,第二次试验时则接受T的一个剂量,如此调换交叉进行,可以在同一动物身上进行不同试品、不同剂量的比较,以去除动物间差异对实验误差的影响,提高实验精确度,节约实验动物。 (2.2)法S和T各两组剂量,用双交叉设计,将动物分成四组;对各组中的每一只动物都标上识别号。每一只动物都按给药次序表进行两次实验。
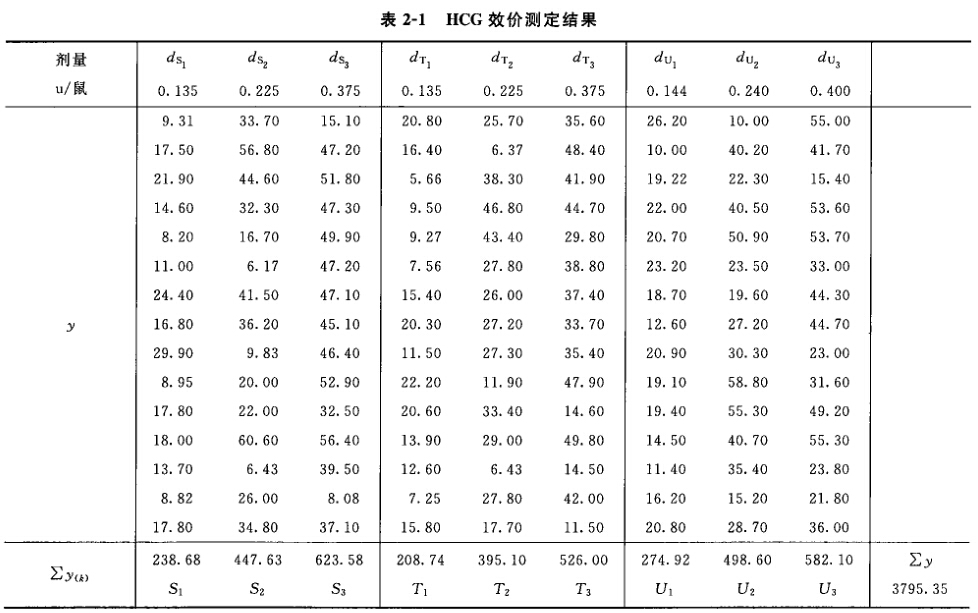
2.平行线测定法的方差分析和可靠性测验 随机设计和随机区组设计的方差分析和可靠性测验 (1)将反应值或其规定的函数(y)按S和T的剂量分组列成方阵表 见表二。
方阵中,K为S和T的剂量组数和,m为各剂量组内y的个数,如为随机区组设计,m为行间或组内所加的因级限制;n为反应的总个数,n=mK。 (2)特异反应剔除和缺项补足 特异反应剔除 在同一剂量组内的各个反应中,如出现个别特大或特小的反应,应按下法判断其是否可以剔除。


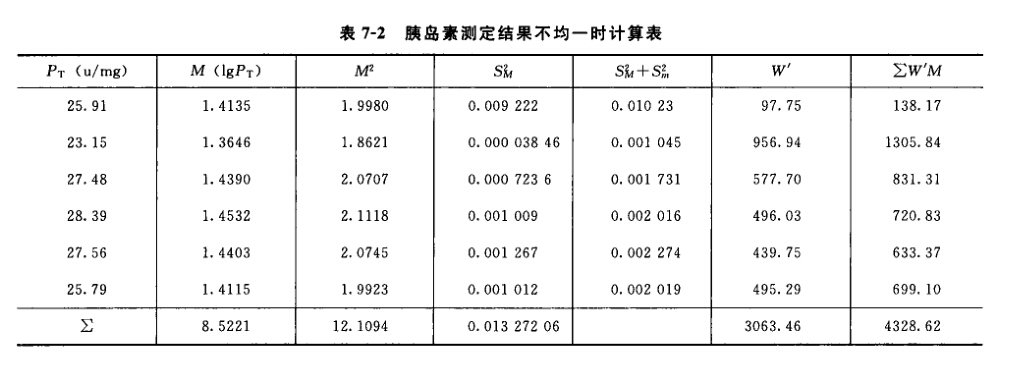
缺项补足 因反应值被剔除或因故反应值缺失造成缺项,致m不等时,根据实验设计类型做缺项补足,使各剂量组的反应个数m相等。 随机设计 对缺失数据的剂量组,以该组的反应均值补入,缺1个反应补1个均值,缺2个反应补2个均值。 随机区组设计 按(13)式计算,补足缺项。
如果缺1项以上,可以分别以y1、y2、y3等代表各缺项,然后在计算其中之一时,把其他缺项y直接用符号y1、y2等当作未缺项代入(1主与缺项数相同的方程组,解方程组即得。 随机区组设计,当剂量组内安排的区组数较多时,也可将缺项所在的整个区组除去。 随机设计的实验结果中,如在个别剂量组多出1~2个反应值,可按严格的随机原则去除,使各剂量组的反应个数m相等。 不论哪种实验设计,每补足一个缺项,就需把s2的自由度减去1,缺项不得超过反应总个数的5%。 (3)方差分析 方阵表(表二)的实验结果,按 (14)~(21)式计算各项变异的差方和、自由度(f)及误差项的方差(s2)。 随机设计 按(14)式、(15)式计算差方和(总)、差方和(剂间)。按(20)式计算差方和(误差)。按(18)式或(21)式计算s2。 随机区组设计 按(14)~(17)式计算差方和(总)、差方和(剂间)、差方和(区组间)、差方和(误差)。按(18)式或(19)式计算s2。

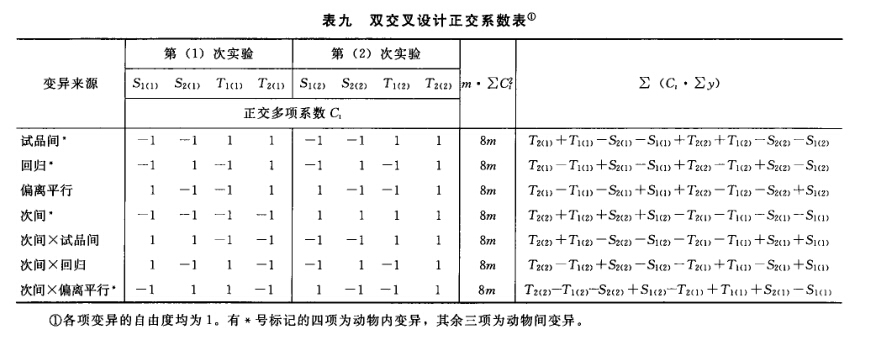
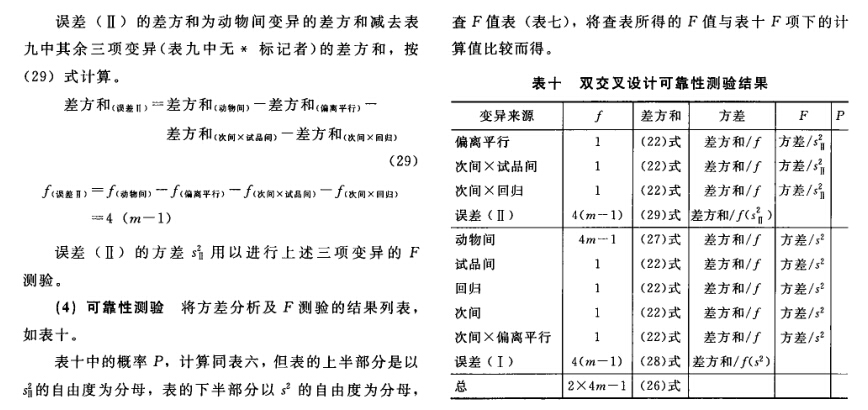
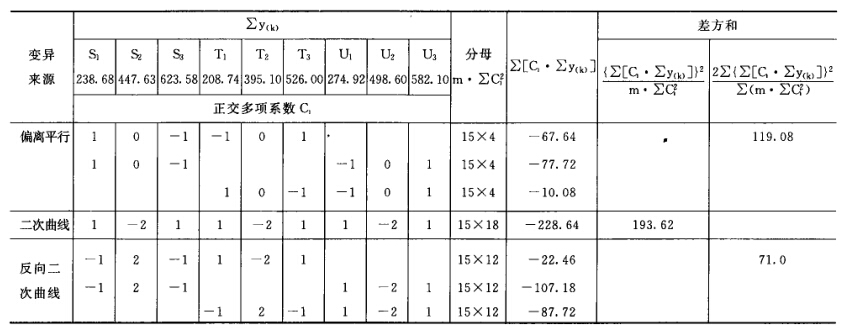
(4)可靠性测验 通过对剂间变异的分析,以测验S和T的对数剂量和反应的关系是否显著偏离平行直线。(2.2)法和(2.2.2)法的剂间变异分析为试品间、回归、偏离平行三项,其他(k.k)法还需再分析二次曲线、反向二次曲线等。 可靠性测验的剂间变异分析

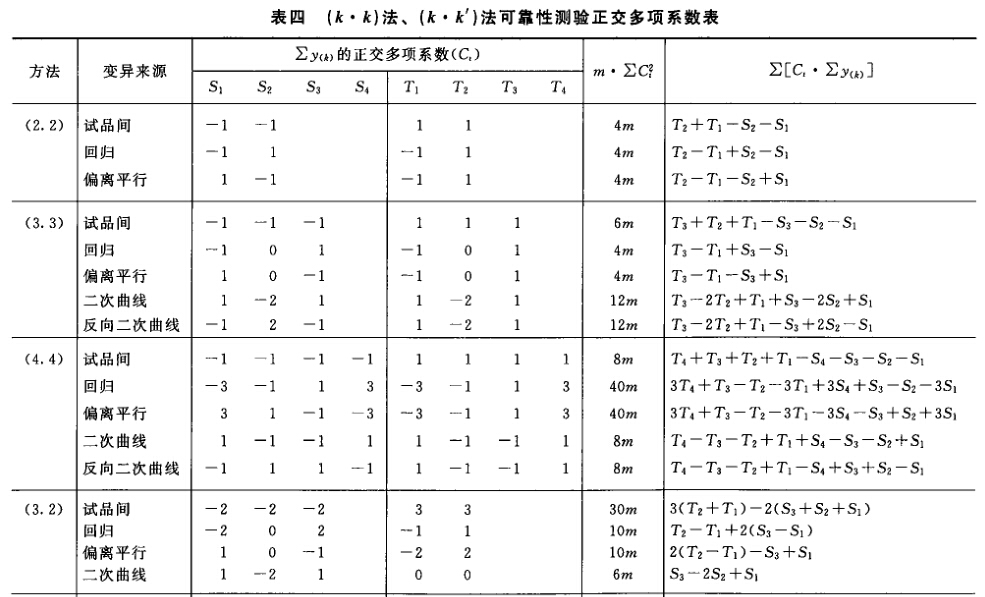
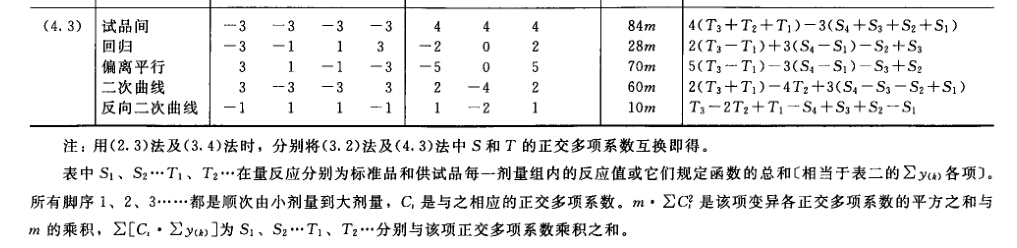

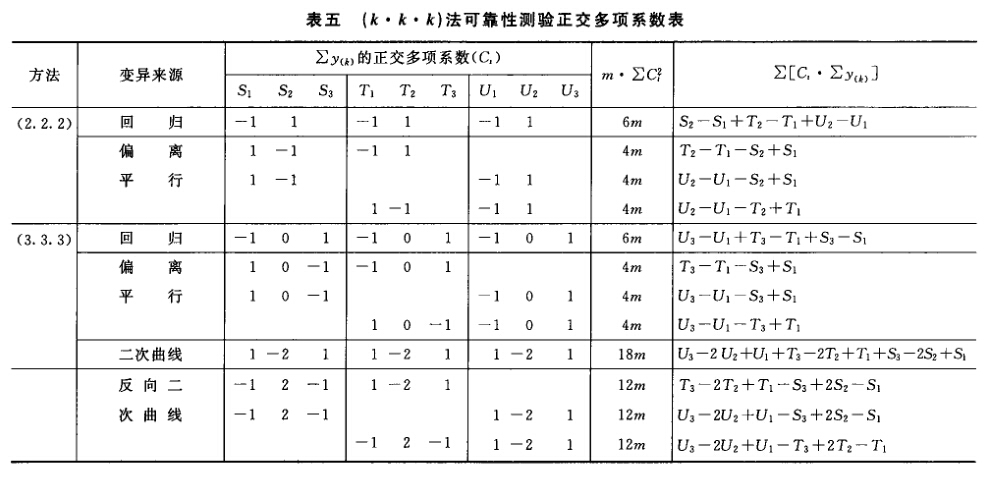
按(18)式计算各项变异的方差。 将方差分析结果列表进行可靠性测验。例如随机区组设计(3.3)法可靠性测验结果列表,见表六。
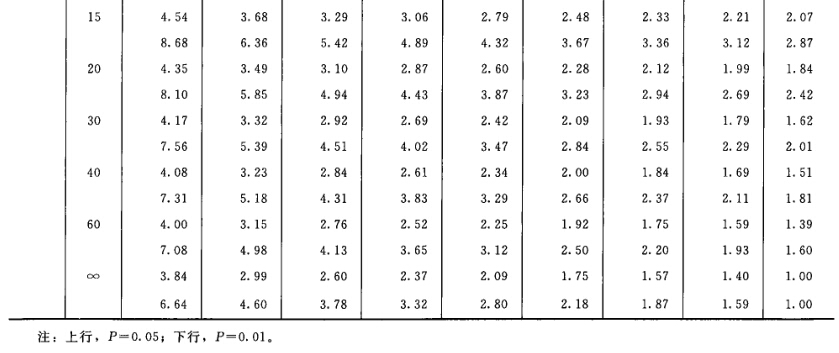
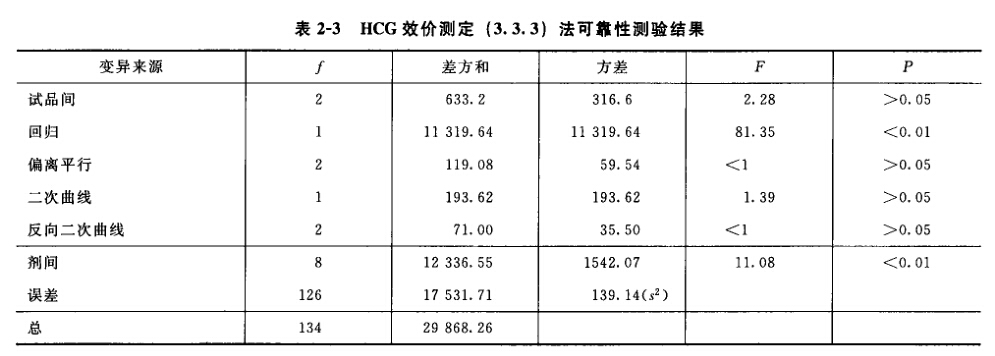
表六中概率P是以该变异项的自由度为分子,误差项(s2)的自由度为分母,査F值表(表七),将查表所得F值与表六F项下的计算值比较而得。当F计算值大于P=0.05或P=0.01的查表值时,则P<0.05或P<0.01,即为在此概率水平下该项变异有显著意义。 随机设计没有区组间变异项。 可靠性测验结果判断 可靠性测验结果,回归项应非常显著(P<0.01)。 (2.2)法、(2.2.2)法偏离平行应不显著(P>0.05)。 其他(k.k)法、(k.k.k)法偏离平行、二次曲线、反向二次曲线各项均应不显著(P>0.05)。 试品间一项不作为可靠性测验的判断标准,试品间变异非常显著者,重复试验时,应参考所得结果重新估计T的效价或重新调整剂量试验。 双交叉设计的方差分析和可靠性测验 (1)双交叉设计实验结果的方阵表 将动物按体重随机分成四组,各组的动物数(m)相等,四组的动物总数为4m。对四组中的每一只动物都加以识别标记,按双交叉设计给药次序表进行实验,各组的每一只动物都给药两次,共得2×4m个反应值。将S、T各两个剂量组两次实 验所得反应值排列成表,见表八。
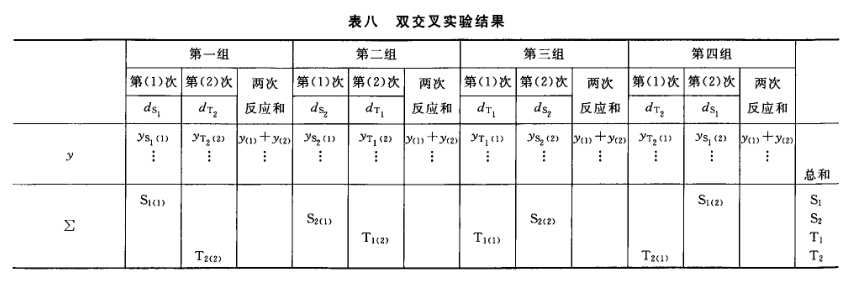





续表



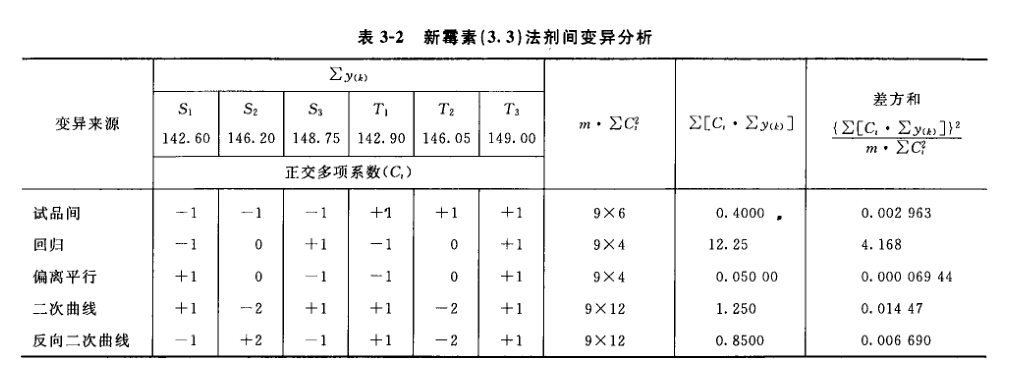
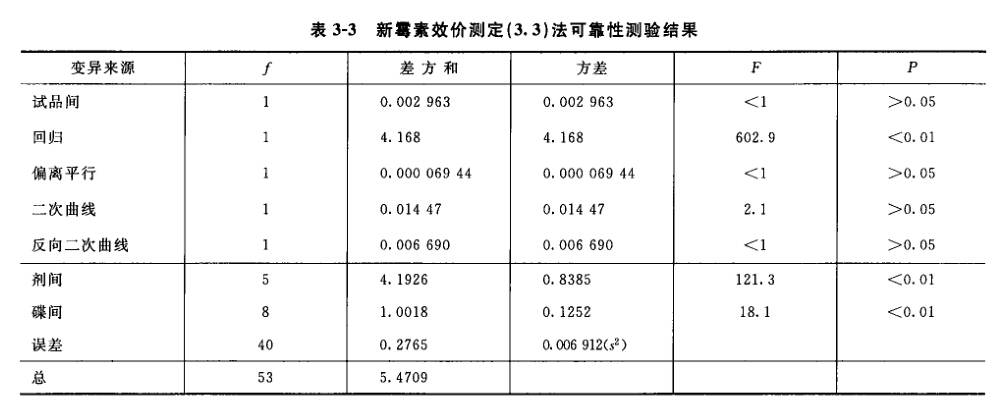
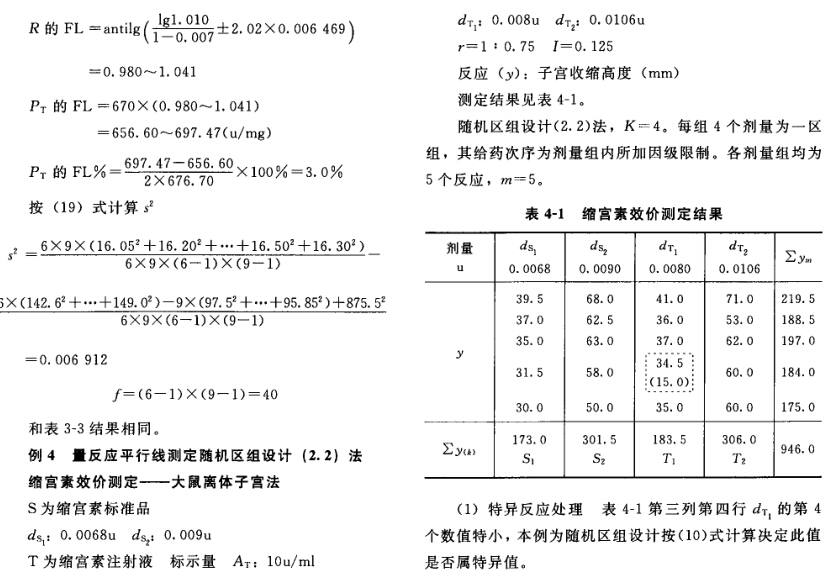

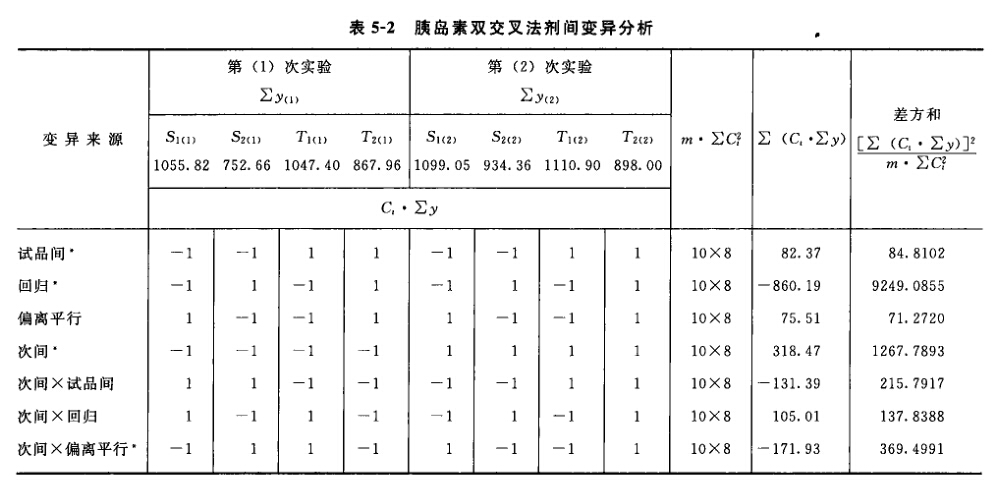
(3)剂间变异分析及可靠性测验 按表四(2.2)法计算,结果见表4-2、表4-3。
结论:回归非常显著(P<0.01),偏离平行不显著(P>0.05),实验结果成立。 区组间差异显著(P<0.05),分离区组间变异,可以减小实验误差。 缩宫素离体子宫效价测定,如区组间变异不显著,也可以不分离区组间变异,用随机设计方差分析法计算。

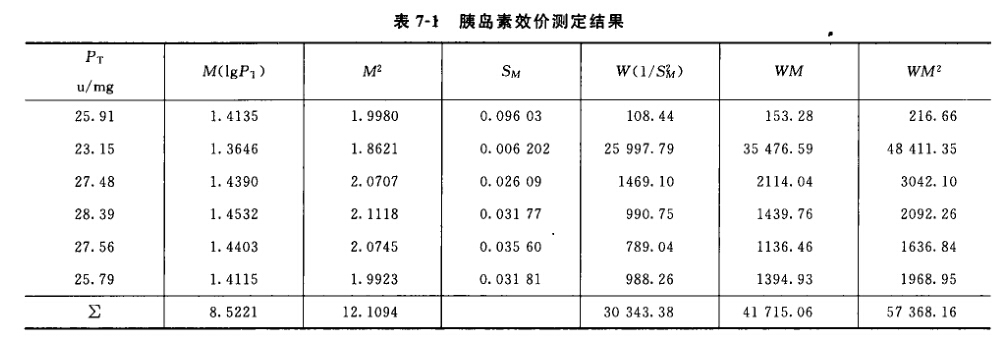
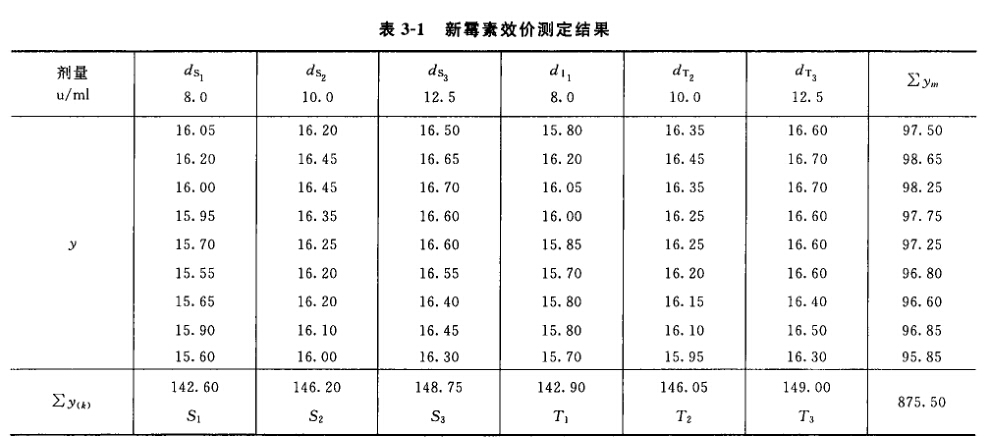
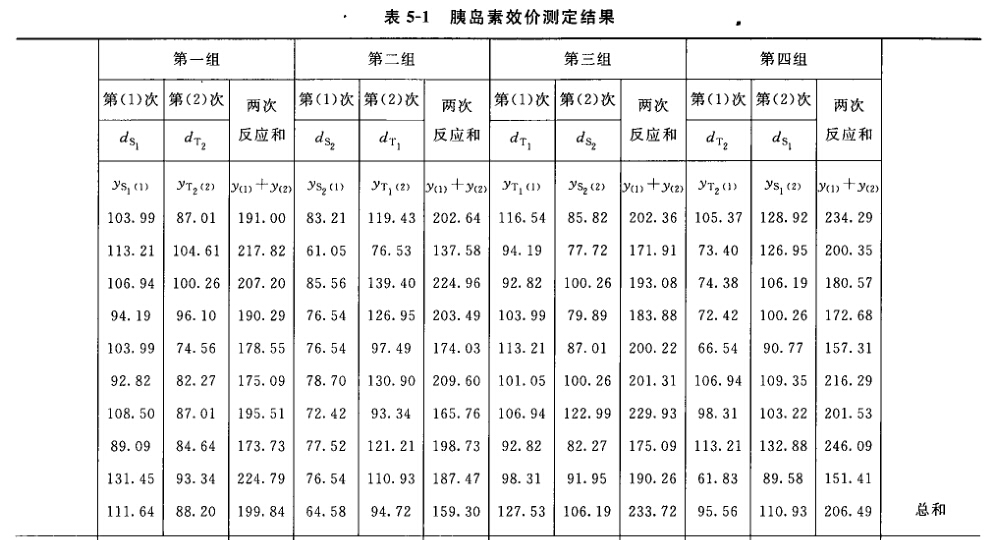
例5 量反应平行线测定(2.2)法双交叉设计胰岛素效价测定----小鼠血糖法 S为胰岛素标准品




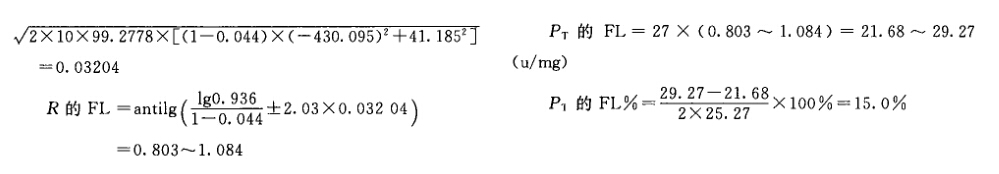
四、实验结果的合并计算 同一批供试品重复n次测定,所得n个测定结果,可用合并计算的方法求其效价PT的均值及其FL。 参加合并计算的n个结果应该是: (1)各个实验结果是独立的,完整的,是在动物来源、实验条件相同的情况下,与标准品同时比较所得的检定结果(PT)。 (2)各次检定结果,经用标示量或估计效价(AT)校正后,取其对数值(lgPT)参加合并计算。


(1)如为个别实验结果影响n次实验结果的均一性,可以剔除个别结果,将其余均一的结果按以上公式进行合并计算,但剔除个别结果应符合“特异反应剔除”的要求。